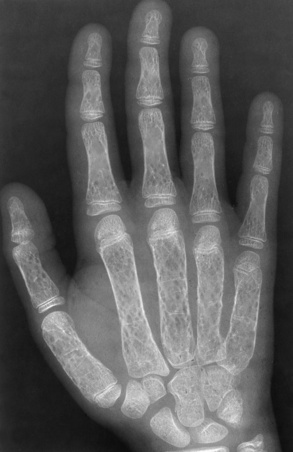
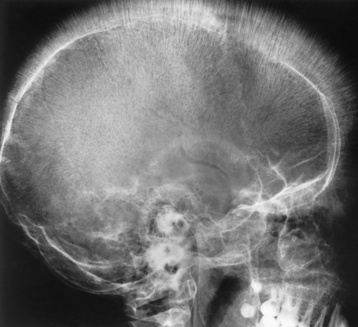
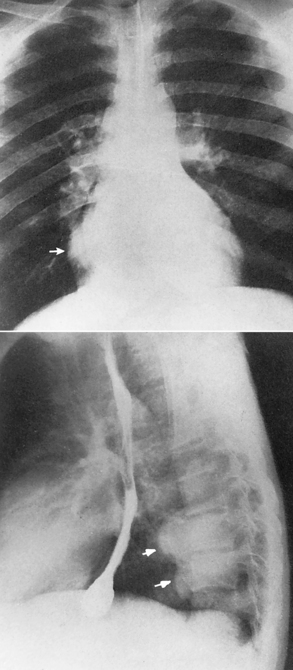
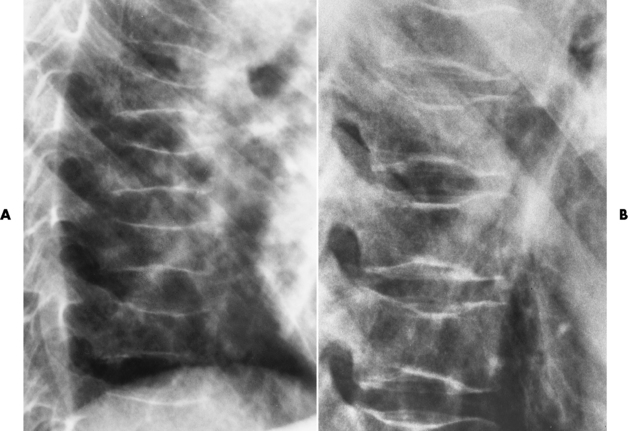
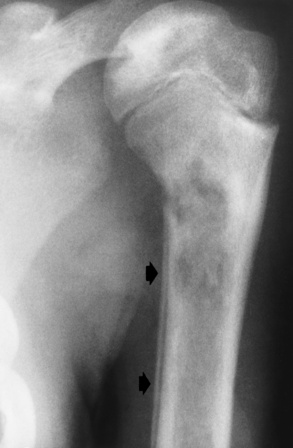
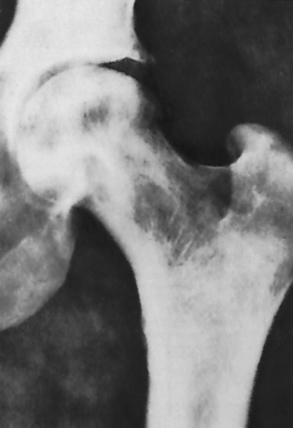
Chapter 9 After reading this chapter, the reader will be able to: 1 Define and describe all bold-faced key terms in this chapter 2 Describe the physiology of the hematopoietic system 3 Identify the basic blood structures on diagrams 4 Differentiate the various pathologic conditions affecting the hematopoietic system as well as their radiographic manifestations Red bone marrow (found in vertebrae, proximal femurs, and flat bones such as the sternum, ribs, skull, and pelvis) and lymph nodes are the blood-forming tissues of the body. Red blood cells (erythrocytes) and platelets (thrombocytes) (Figure 9-1) are made in red bone marrow, whereas white blood cells (leukocytes) (Figure 9-2) are produced in both red marrow and lymphoid tissue. If a bone marrow puncture is necessary for diagnostic testing, the iliac rim and sternum are good sites. Erythrocytes are biconcave disks without a nucleus that contain hemoglobin, an iron-based protein that carries oxygen from the respiratory tract to the body’s tissues. In a normal person, there are 4.5 to 6 million red blood cells in each cubic millimeter of blood. The amount of hemoglobin per deciliter is approximately 14 g in women and 15 g in men. Leukocytes, or white blood cells, normally number from 5000 to 10,000/mm3 of blood (see Figure 9-2). Unlike erythrocytes, there are several types of white blood cells. Neutrophils (polymorphonuclear leukocytes), which make up 55% to 75% of white blood cells, defend the body against bacteria by ingesting these foreign organisms and destroying them (phagocytosis). The number of polymorphonuclear leukocytes in the blood increases enormously in acute infections because the bone marrow rapidly releases into the bloodstream the large numbers of these cells kept in reserve. Eosinophils (1% to 4%) are red-staining cells whose number greatly increases in allergic and parasitic conditions. The third type of leukocyte is the basophil (0% to 1%), which contains granules that stain blue. These three types of cells are formed in the sinusoids of bone marrow, and they, like red blood cells, go through immature stages before reaching the adult form. The final type of white blood cell is the monocyte, which is actively phagocytic and plays an important part in the inflammatory process. Monocytes are formed in the bone marrow and represent about 2% to 8% of white blood cells. Platelets, the smallest blood cells, are essential for blood clotting (see Figure 9-1). Normally there are about 150,000 to 400,000 platelets in every cubic millimeter of blood. Anemia refers to a decrease in the amount of oxygen-carrying hemoglobin in the peripheral blood. This reduction can be attributable to improper formation of new red blood cells, an increased rate of red blood cell destruction, or a loss of red blood cells as a result of prolonged bleeding. Regardless of the cause, a hemoglobin deficiency causes the anemic person to appear pale. This is best appreciated in the mucous membranes of the mouth and conjunctiva, and in the nail beds. A decrease in the oxygen-carrying hemoglobin impairs the delivery of an adequate oxygen supply to the cells and tissues, leading to fatigue and muscular weakness and often to shortness of breath on exertion (dyspnea). To meet the body’s need for more oxygen, the respiratory rate increases and the heart beats more rapidly. Spherocytosis, sickle cell anemia, and thalassemia are the major hereditary hemolytic anemias. In spherocytosis the erythrocytes have a circular rather than a biconcave shape, making them fragile and susceptible to rupture. In sickle cell anemia, which is generally confined to African Americans, the hemoglobin molecule is abnormal and the red blood cells are crescentic or sickle shaped and tend to rupture. A defect in hemoglobin formation is also responsible for thalassemia, which occurs predominantly in persons living near the Mediterranean Sea, especially those of Italian, Greek, or Sicilian descent. Radiographic Appearance: The hemolytic anemias produce a variety of radiographic abnormalities. Although the radiographic findings are similar in the various types of hemolytic anemia, they tend to be most severe in thalassemia and least prominent in spherocytosis. Extensive marrow hyperplasia, the result of ineffective erythropoiesis and rapid destruction of newly formed red blood cells, causes generalized osteoporosis with pronounced widening of the medullary spaces and thinning of the cortices in long and tubular bones (Figure 9-3). As the fine secondary trabeculae are resorbed, new bone is laid down on the surviving trabeculae, thickening them and producing a coarsened pattern. Normal modeling of long bones does not occur because the expanding marrow flattens or even bulges the normally concave surfaces of the shafts. In the skull, there is widening of the diploic space and thinning or complete obliteration of the outer table. When the hyperplastic marrow perforates or destroys the outer table, it proliferates under the invisible periosteum, and new bone spicules are laid down perpendicular to the inner table. This produces the characteristic hair-on-end appearance of vertical striations in a radial pattern (Figure 9-4). Extramedullary hematopoiesis is a compensatory mechanism of the reticuloendothelial system (liver, spleen, lymph nodes) in patients with prolonged erythrocyte deficiency resulting from the destruction of red blood cells or the inability of normal blood-forming organs to produce them. Paravertebral collections of hematopoietic tissue may appear on chest radiographs as single or multiple, smooth or lobulated, posterior mediastinal masses that are usually located at the lower thoracic levels (Figure 9-5). In sickle cell anemia, expansile pressure of the adjacent intervertebral disks produces characteristic biconcave indentations on both the superior and inferior margins of the softened vertebral bodies, giving the appearance of fish vertebrae (Figure 9-6, A). Another typical appearance is the result of the development of localized steplike central depressions of multiple vertebral end plates (Figure 9-6, B). This is most often caused by circulatory stasis and ischemia, which retard growth in the central portion of the vertebral cartilaginous growth plate. The periphery of the growth plate, which has a different blood supply, continues to grow at a more normal rate. Acute osteomyelitis, often caused by Salmonella infection, is a common complication in sickle cell disease. The resulting lytic destruction and periosteal reaction may be extensive, often involving the entire shaft and multiple bones (Figure 9-7). Radiographically, it may be impossible to distinguish between osteomyelitis and bone infarction without infection (Figure 9-8). Renal abnormalities can be demonstrated by excretory urography in about two thirds of patients with sickle cell disease. A serious complication is renal papillary necrosis (see Figure 6-21), which is probably related to vessel obstruction within the papillae and may produce sinuses or cavity formation within one or more papillae.
Hematopoietic System
Physiology of the blood
Diseases of red blood cells
Hemolytic Anemia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Endocrine System