FIGURE 6-1. Homeostatic mechanisms involved in sodium, potassium, and water balance.
Changes in body water and plasma volume can directly or indirectly affect the serum sodium concentration. For example, as the result of changes in effective circulating volume, baroreceptors and osmoreceptors will respond accordingly in an attempt to restore an isovolemic state of the body. Baroreceptors are located in the carotid sinus, aortic arch, cardiac atria, hypothalamus, and the juxtaglomerular apparatus in the kidney. Stimulation of these receptors will promote urinary loss of water and sodium. Osmoreceptors are present primarily in the hypothalamus. The three major effectors in response to the stimulation of the osmoreceptors include vasopressin or antidiuretic hormone (ADH), the renin-angiotensin-aldosterone system (RAAS), and natriuretic peptides. The resultant renal effects from these three distinct pathways will alter the homeostasis of water and sodium.
The kidneys are the primary organ responsible for the retention and excretion of body sodium and water. The glomeruli receive and filter about 180 L of plasma and 600 g (nearly 26,000 mEq) of sodium per day. On average, less than 2 L of water and between 0.1–40 g of sodium are excreted in the urine, depending on the fluid status of the individual. While almost 100% of the plasma sodium is filtered by the glomeruli, less than 1% is excreted in the urine under normal circumstances. The proximal tubule and the loop of Henle each account for approximately 45% of sodium reabsorption.
The homeostatic mechanism for water and sodium also involves the equilibrium among intravascular, interstitial, and intracellular fluids.3 Net movement of water occurs from areas of low osmolality to areas of high osmolality. This effect can be readily observed in patients with a low serum osmolality due to a deficit of serum sodium or excess of plasma water. Water then moves from the plasma compartment to the higher osmolality in the interstitial space to minimize the osmolar gap.3 In the presence of high hydrostatic and oncotic pressure gaps across capillary walls, the net effect is excessive interstitial water accumulation and edema formation.3,4
Antidiuretic hormone (vasopressin). Antidiuretic hormone (ADH), also known as arginine vasopressin, is a nonapeptide hormone that regulates renal handling of free water exclusively. By altering the amount of water reabsorbed by the kidney, ADH has a pivotal effect in changing or maintaining serum sodium concentration, and it is secreted by the magnocellular neurons in the supraoptic and paraventricular nuclei of the hypothalamus, where both osmoreceptors and baroreceptors are present to detect vascular fluid changes. Its release is stimulated by (1) hypovolemia (detected by baroreceptors), (2) thirst, (3) increased serum osmolality, and (4) angiotensin II. The plasma half-life of ADH is 10–20 minutes, and it is rapidly deactivated and eliminated by the liver, kidneys, and a plasma enzyme called vasopressinase.
Antidiuretic hormone decreases urine output by augmenting the permeability of the collecting tubules to increase the net reabsorption of water. Circulating serum ADH binds to type 2 vasopressin (V2) receptors in the collecting tubule, which in turn stimulates the formation of a water channel, known as aquaporin-2. Aquaporin-2 facilitates the reabsorption of water from the lumen back into the renal blood supply in the systemic circulation, causing a decrease in diuresis. However, if serum sodium is high but blood volume is normal (e.g., normovolemia with hyperosmolality), the effect from the baroreceptors overrides the further release of ADH, thus preventing volume overload (i.e., hypervolemia).3
In patients with the syndrome of inappropriate ADH (SIADH) secretion, an abnormally high quantity of ADH is present in the systemic circulation. This condition results in increased water reabsorption, which could cause a dilutional effect in serum sodium. In conjunction with an increased free water intake, a low serum sodium concentration is commonly observed in these patients. Urine osmolality and urine electrolyte concentrations are often increased due to the decreased urinary excretion of free water. Conversely, central diabetes insipidus (DI) occurs when hypothalamic ADH synthesis or release is impaired. Patients with DI commonly present with hypernatremia due to the increased renal wasting of free water. In some cases, the kidneys fail to respond to normal or high quantities of circulating ADH. This condition is called nephrogenic diabetes insipidus. In central or nephrogenic DI, little water reabsorption occurs, resulting in a large urine output with low urine osmolality.3 (Chapter 8: The Kidneys offers an in-depth discussion of the effects of other diseases on urine composition.)
Drugs may affect ADH release or action, which may exacerbate SIADH or DI. Increased ADH release has been reported with chlorpropamide, tolbutamide, cyclophosphamide, carbamazepine, oxcarbazepine, some opiate derivatives, clofibrate, oxytocin, vincristine, phenothiazines, some tricyclic antidepressants, and a number of serotonin reuptake inhibitors. Because of their ability to increase ADH release, some of these drugs are used in the treatment of central DI. In contrast, other drugs that decrease ADH release (e.g., demeclocycline or lithium carbonate) have been used to treat SIADH. Selective type-2 vasopressin receptor (V2) antagonists (e.g., tolvaptan, conivaptan) modulate the renal handling of water and have a direct effect on sodium homeostasis.
Renin-angiotensin-aldosterone system. Renin is a glycoprotein that catalyzes the conversion of angiotensinogen to angiotensin I, which is further converted to angiotensin II primarily in the lungs. However, angiotensin II can also be formed locally in the kidneys. Angiotensin II, a potent vasoconstrictor, is important in maintaining optimal perfusion pressure to end organs especially when plasma volume is decreased. In addition, it induces the release of aldosterone, ADH, and, to a lesser extent, cortisol.
Aldosterone is a hormone with potent mineralocorticoidal actions. It affects the distal tubular reabsorption of sodium rather than water.4 This hormone is released from the adrenal cortex. Besides angiotensin II, various dietary and neurohormonal factors including low serum sodium, high serum potassium, and low blood volume can also stimulate its release. Aldosterone acts on renal Na-K-ATPase to increase urinary excretion of potassium from the distal tubules in exchange for sodium reabsorption. Because of its effect on renal Na/K exchange, aldosterone has a profound effect on serum potassium, while its effect on serum sodium is relatively modest. As serum sodium increases, so does water reabsorption, which follows the osmotic gradient.3 Renal arteriolar blood pressure (BP) then increases, which helps maintain the glomerular filtration rate (GFR). Ultimately, more water and sodium pass through the distal tubules, overriding the initial effect of aldosterone.3,4
Natriuretic peptides. Atrial natriuretic factor (ANF), also known as atrial natriuretic peptide, is a vasodilatory hormone synthesized and primarily released by the right atrium. It is secreted in response to plasma volume expansion, as a result of increased atrial stretch. Atrial natriuretic factor inhibits the juxtaglomerular apparatus, zona glomerulosa cells of the adrenal gland, and the hypothalamus-posterior pituitary. As a result, a global down regulation of renin, aldosterone, and ADH, respectively, is achieved. Atrial natriuretic factor directly induces glomerular hyperfiltration and reduces sodium reabsorption in the collecting tubule. A net increase in sodium excretion is achieved. Therefore, ANF can decrease serum and total body sodium. Brain natriuretic peptide (BNP) is produced and secreted primarily by the ventricles in the brain, and to a much smaller extent, the atrium. Similar to ANP, BNP also regulates natriuretic, endocrine, and hemodynamic responses and may affect sodium homeostasis. An increase in blood volume or pressure, such as chronic heart failure and hypertension, enhances BNP secretion, which induces a significant increase in natriuresis and to a lesser extent, urinary flow (i.e., diuresis). Plasma BNP concentrations are found to correlate with the magnitude of left ventricular heart failure and the clinical prognosis of these patients.
Hyponatremia is loosely defined as a serum sodium concentration less than 136 mEq/L (<136 mmol/L). Hyponatremia may occur when total body water status is low (i.e., dehydration), normal, or high (i.e., fluid overload). Therefore, natremic status cannot be assessed without first assessing the fluid and water status of the patient. Fluid status should be evaluated based on vital signs; other supportive laboratory findings if available (e.g., serum, blood urea nitrogen–creatinine ratio, hematocrit to hemoglobin concentration ratio, or urine electrolyte assessment); recent changes in body weight; recent medical, surgical, and nutrition history; and findings from the physical examination. More importantly, the patient’s renal function, hydration status, and fluid intake and output must be carefully assessed and closely monitored. The most common causes of hyponatremia can be broken down into two types: sodium depletion in excess of total body water loss (e.g., severe dehydration with true depletion of total body sodium) and dilutional hyponatremia (i.e., water intake greater than water output). Dilutional hyponatremia can be further categorized into five subtypes: (1) primary dilutional hyponatremia (e.g., SIADH and renal failure); (2) neuroendocrine (e.g., adrenal insufficiency and myxedema); (3) psychiatric disorder (e.g., psychogenic polydipsia); (4) osmotic hyponatremia (e.g., severe hyperglycemia); and (5) thiazide diuretic-induced.
Most patients with hyponatremia remain asymptomatic until serum sodium approaches 120 mEq/L. Infusion of hypertonic saline (e.g., 3% NaCl solution) is usually not necessary unless serum sodium concentration is less than 120 mEq/L, altered mental status is present, and if the patient is fluid restricted (e.g., heart failure, chronic renal failure). As with most electrolyte disorders, the chronicity of the imbalance is a major determinant of the severity of signs and symptoms. For example, hyponatremia in patients with congestive heart failure (CHF) secondary to chronic, progressive volume overload and decreased renal perfusion is less likely to be symptomatic than a patient who is hyponatremic due to rapid infusion of a hypotonic solution. The most commonly reported symptom associated with hyponatremia is altered mental status (List 6-1). If serum sodium continues to fall, cerebral edema can worsen and intracranial pressure will continue to rise. More severe symptoms such as seizure, coma, and, subsequently, death may result.3–6
| LIST 6-1. Signs and Symptoms of Hyponatremia |
| Agitation |
| Anorexia |
| Apathy |
| Depressed deep-tendon reflexes |
| Disorientation |
| Hypothermia |
| Lethargy |
| Muscle cramps |
| Nausea |
| Seizures |
Hyponatremia associated with total body sodium depletion. Hyponatremia associated with low total body sodium reflects a reduction in total body water, with an even larger reduction in total body sodium. This condition is primarily caused by depletion of extracellular fluid, which stimulates ADH release to increase renal water reabsorption even at the expense of causing a transient hypo-osmotic state. Some common causes include vomiting; diarrhea; intravascular fluid losses due to burn injury and pancreatitis; Addison disease; and certain forms of renal failure (e.g., salt-wasting nephropathy).3 This type of hyponatremia may also occur in patients treated too aggressively with diuretics who receive sodium-free solutions as replacement fluid.
Low serum sodium can also result when a large quantity of an osmotically active substance enters the bloodstream, resulting in a dilutional hyponatremia.3 This situation can occur with the use of mannitol, an osmotic diuretic agent, as well as in the presence of hyperglycemia. In hyperglycemia, the elevated serum glucose concentration results in high serum osmolarity, thus creating an osmolar gradient between the plasma compartment and the extracellular fluid leading to a shift of water into the intravascular space. The net effect is a dilution of the serum sodium concentration resulting in hyponatremia.3,5 In the absence of other causes impairing sodium homeostasis, the effect should be transient and should be reversed once serum glucose is normalized. However, hyperglycemia also leads to fluid loss through an osmotic diuretic effect. Hence, dilutional hyponatremia occurs as long as the rate of water moving from the cells into the blood is greater than the volume of water excreted through the urine. As cellular water diminishes and diuresis continues, plasma sodium may increase progressively.3,5
Patients with hyponatremia associated with low total body sodium often exhibit signs and symptoms of dehydration. These manifestations include thirst, dry mucous membranes, weight loss, sunken eyes, diminished urine output, and diminished skin turgor.3
Hyponatremia associated with normal total body sodium. Also called euvolemic or dilutional hyponatremia, this condition refers to impaired water excretion without any alteration in sodium excretion. Etiologies include any mechanism that enhances ADH secretion or potentiates its action at the collecting tubules. This condition can occur as a result of glucocorticoid deficiency, severe hypothyroidism, and administration of water to a patient with impaired water excretion capacity.3,5 Syndrome of inappropriate ADH commonly results in hyponatremia, secondary to continued ADH secretion despite low serum osmolality.
Impaired ADH response can be precipitated by many factors, including medications (Figure 6-2). Syndrome of inappropriate ADH has been reported in patients with certain tumors, such as lung cancer, pancreatic carcinoma, thymoma, and lymphoma. Antidiuretic hormone release from the parvicellular and magnocellular neurons may be stimulated by cytokines such as interleukin (IL-2, IL-6, IL-1 beta), and tumor necrosis factor (TNF-a). Likewise, head trauma, subarachnoid hemorrhage, hydrocephalus, Guillain-Barré syndrome, pulmonary aspergillosis, and occasionally tuberculosis may increase hypothalamic ADH production and release. Patients with SIADH produce concentrated urine with high urine osmolality (usually >200 mOsm/kg H2O) and urine sodium excretion (as reflected in a urine sodium concentration that is usually >20 mEq/L). They have normal renal, adrenal, and thyroid function and have no evidence of volume abnormalities.3,5
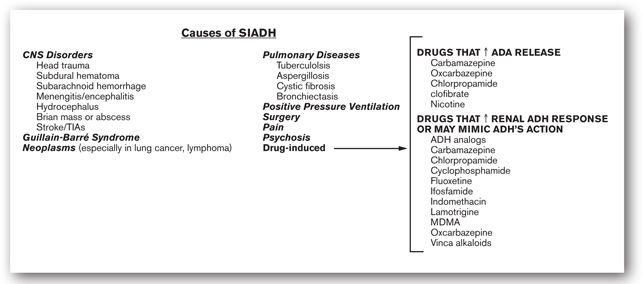
FIGURE 6-2. Etiology of SIADH.
Hyponatremia may also be associated with an increase of total body sodium. This condition implies an increase in total body sodium with an even larger increase in total body water. It is frequently observed in edematous states such as CHF, cirrhosis, nephrotic syndrome, and chronic kidney disease (CKD). In these patients, renal handling of water and sodium is usually impaired.3,5
Tests for Assessing Fluid Status
Fractional Excretion of Sodium (FENa)
Normal range: 1% to 2%
In most cases, natremic disorders cannot be effectively managed without first optimizing the overall fluid status. Therefore, when a serum sodium value is abnormal, the clinician should first evaluate whether vascular volume is optimal. In addition to physical examinations and history, FENa may help validate these findings, especially in patients whose physical examination results may be limited by other confounders (e.g., the use of antihypertensive drugs, heart failure). The FENa may be determined by the use of a random urine sample to determine renal handling of sodium. FENa, the measure of the percentage of filtered sodium excreted in the urine, can be calculated using the following equation:
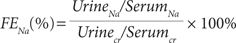
Values greater than 2% usually suggest that the kidneys are excreting a higher than normal fraction of the filtered sodium, implying likely renal tubular damage. Conversely, FENa values less than 1% generally imply preservation of intravascular fluid through renal sodium retention, suggesting prerenal causes of renal dysfunction (e.g., hypovolemia and cardiac failure). Since acute diuretic therapy can increase the FENa to 20% or more, urine samples should be obtained at least 24 hours after diuretics have been discontinued.3 Minicase 1 demonstrates how to calculate FENa.
A Case of Hyponatremia
JANE W., A 74-YEAR-OLD WOMAN, had a history of coronary artery disease, CHF, and type 2 diabetes mellitus. She was brought to the emergency department by her daughter, who complained about her mother’s increasing disorientation over the past week. Medications (all taken orally) at the time of admission were benazepril 10 mg daily, digoxin 0.125 mg daily, furosemide 40 mg twice a day, metformin extended-released 2 g daily, sitagliptin 100 mg daily, and acetaminophen 650 mg PRN. She last took these medications the morning before presentation to the hospital. A review of systems revealed lethargy and apathy with no apparent distress. Vital signs showed a BP of 110/75 mm Hg at supine position (standing BP 105/70 mm Hg), a regular heart rate at 96 beats per minute (standing 100 beats per minute), and a rapid and shallow respiratory rate of 36 breaths per minute. Her laboratory findings were unremarkable, except for serum sodium of 120 mEq/L (136–142 mEq/L) and serum glucose of 185 mg/dL (70–110 mg/dL).
Question: What subjective and objective findings in Jane W. are consistent with the presentation of hyponatremia? What are the potential etiologies of hyponatremia in Jane W.?
Discussion: Aside from her low serum sodium concentration, her other major symptoms consistent with hyponatremia are mostly CNS-related symptoms, such as lethargy and apathy. Rapid shallow breathing (Cheyne-Stokes respiration) may be related to severe heart failure or an altered vascular fluid status. A clinician must determine volume status when assessing a patient with sodium abnormality. To further determine possible etiologies, the clinician should assess Jane W.’s body fluid status. The fact that she had normal blood pressure and a lack of orthostatic hypotension with essentially no change in heart rate suggests that hypovolemia is unlikely. Therefore, the primary cause of her symptoms at presentation is most likely associated with hyponatremia. Furosemide should be held as it is known to cause increased renal sodium loss which would worsen her current condition. Renal handling of sodium using other laboratory or diagnostic tests such as urine sodium excretion, and urine osmolality and FENa should be considered 24 hours after the discontinuation of furosemide, as the natriuretic action of loop diuretics could limit the accuracy in interpreting these test results.
Blood Urea Nitrogen (BUN): Serum Creatinine (SCr) Ratio
Normal range: <20:1
The BUN: SCr ratio can provide useful information to assess fluid status. When this ratio is higher than 20:1, dehydration is usually present. As extracellular fluid volume is diminished, the kidneys increase their reabsorption of urea but not creatinine. Therefore, BUN increases by a larger magnitude than the SCr concentration in dehydrated individuals, leading to a rise in the BUN: SCr ratio.
Hypernatremia is defined as a serum sodium concentration greater than 142 mEq/L (>142 mmol/L). High serum sodium concentrations are common in patients with either an impaired thirst mechanism (e.g., neurohypophyseal lesion, especially after suffering from a stroke) or an inability to replete water deficit through normal insensible losses (i.e., uncontrollable water loss through respiration or skin) or from renal or GI losses. All hypernatremic states increase serum osmolality. Similar to hyponatremia, hypernatremia may occur in the presence of high, normal, or low total body sodium content.3,4,6
The clinical manifestations of hypernatremia primarily involve the neurological system (List 6-2). These manifestations are the consequence of cellular dehydration, particularly in the brain. In adults, acute elevation in serum sodium above 160 mEq/L (>160 mmol/L) is associated with 75% mortality rate. Unfortunately, neurological sequelae are common even in survivors. In order to assess the etiology of hypernatremia, it is important to determine (1) urine production; (2) sodium intake; and (3) renal solute concentrating ability, which reflects ADH activity.
| LIST 6-2. Signs and Symptoms of Hypernatremia | |
| Thirst | |
| Restlessness | |
| Irritability | |
| Lethargy | |
| Muscle twitching | |
| Seizures | |
| Hyperreflexia | |
| Coma | |
| Death |
Hypernatremia associated with low total body sodium occurs when the loss of water exceeds the loss of sodium.3 The thirst mechanism generally increases water intake, but this adjustment is not always possible (e.g., institutionalized elderly patients). This condition may also be iatrogenic when hypotonic fluid losses (e.g., profuse sweating and diarrhea) are replaced with an inadequate quantity of water and salt. In these circumstances, fluid loss should be replaced with intravenous (IV) dextrose solutions or hypotonic saline solutions.3,5 In hypernatremic patients presenting with high urine osmolality (>800 mOsm/L, roughly equivalent to a specific gravity of 1.023) and low urine sodium concentrations (<10 mEq/L), these laboratory results reflect an intact renal concentrating mechanism. Signs and symptoms of dehydration should be carefully examined. These include orthostatic hypotension, flat neck veins, tachycardia, poor skin turgor, and dry mucous membranes. In addition, the BUN: SCr ratio may be greater than 20 secondary to dehydration.3,5
Hypernatremia may be associated with normal total body sodium, also known as euvolemic hypernatremia. This condition refers to a general water loss without concurrent sodium loss.3 Because of water redistribution between the intracellular and extracellular fluid, no plasma volume contraction is usually evident unless water loss is substantial (Minicase 2). Etiologies include increased insensible water loss (e.g., fever, extensive burns, and mechanical ventilation) and central and nephrogenic DI. The clinician should be aware of drugs that may cause nephrogenic DI (List 6-3).3,5
A Case of Hypernatremia After a Complete Hypophysectomy
RYAN L., A 21-YEAR-OLD MAN, was admitted to the neurosurgical service for a complete hypophysectomy to remove a pituitary tumor. He was otherwise in good health and was on no chronic medication. His postoperative medications included morphine (administered in a patient-controlled analgesia pump) and IV cefazolin 1 g intravenously every 8 hours.
On the day after surgery, Ryan L.’s urine output reached 4.5 L in 24 hours. His physical exam was unremarkable, and his vital signs were stable. Clinical laboratory tests included serum sodium 152 mEq/L (136–142 mEq/L), potassium 3.8 mEq/L (3.8–5.0 mEq/L), chloride 102 mEq/L (95–103 mEq/L), total carbon dioxide content 24 mmol/L (24–30 mmol/L), glucose 98 mg/dL (70–110 mg/dL), serum phosphate 2.9 mg/dL (2.3–4.7 mg/dL), BUN 18 mg/dL (8–23 mg/dL), and SCr 0.9 mg/dL (0.6–1.2 mg/dL). Urine specific gravity was 1.003 (1.016–1.022). On the following day, his daily urine volume reached 8 L, and he complained of being persistently thirsty despite drinking 14 glasses of water in the past 24 hour period.
Question: What is the primary cause of the sodium disorder in Ryan L.?
Discussion: Surgical procedures that could potentially affect pituitary gland functions are a major risk factor for sodium disorders since the release and regulation of ADH may be affected. Postoperatively, this patient presented with severely elevated urine output with persistent thirst. This suggests that an ADH-related disorder, DI, is likely present with a serious concern for altered sodium homeostasis. His relatively normal vital signs were maintained by his ability to dramatically increase his water intake; otherwise, he likely would have developed hypotension. This is an acute medical problem and the diagnosis should be established quickly with the help of several laboratory tests such as urine sodium, serum sodium, and urine osmolality. The diagnosis of DI can be confirmed with low urine osmolality, low urine sodium. These results, together with his elevated serum sodium and low urine specific gravity, as well as his clinical manifestation of polyuria and polydipsia, are consistent with the diagnosis of DI.
Free water supplementation by mouth or IV fluid administration with dextrose 5% is necessary for correcting hypernatremia and preventing hypovolemia. If the diagnosis of DI is subsequently established, vasopressin or desmopressin, a synthetic analog of vasopressin, will be reasonable option for long-term maintenance therapy. Hypernatremia may be associated with high total body sodium. This form of hypernatremia is the least common, since sodium homeostasis is maintained indirectly through the control of water, and defects in the system usually affect total body water more than total body sodium.3 This form of hypernatremia usually results from exogenous administration of solutions containing large amounts of sodium:
- Resuscitative efforts using hypertonic sodium bicarbonate
- Inadvertent IV infusion of hypertonic saline (i.e., solutions >0.9% sodium chloride)
- Inadvertent dialysis against high sodium-containing solution
- Sea water, near drowning
Primary hyperaldosteronism and Cushing syndrome may also cause this form of hypernatremia. Large quantities of sodium can be found in the urine of these patients. Signs and symptoms include diminished skin turgor and elevated plasma proteins.3,5
Potassium
Normal range: 3.8–5.0 mEq/L or 3.8–5.0 mmol/L
Potassium is the primary cation in the intracellular space, with an average intracellular fluid concentration of about 140 mEq/L (140 mmol/L). The major physiological role of potassium is in the regulation of muscle and nerve excitability. It may also play important roles in the control of intracellular volume (similar to the ability of sodium in controlling extracellular volume), protein synthesis, enzymatic reactions, and carbohydrate metabolism.7,8
Physiology
The most important aspect of potassium physiology is its effect on action potential, especially on muscle and nervous tissue excitability.2 During periods of potassium imbalance, the cardiovascular system is of principal concern. Cardiac muscle cells depend on their ability to change their electrical potentials, with accompanying potassium flux when exposed to the proper stimulus, to result in muscle contraction and nerve conduction.7,8
One important aspect of potassium homeostasis is its distribution equilibrium. In a 70-kg man, the total body potassium content is about 4000 mEq. Of that amount, only 60 mEq is located in the extracellular fluid with the remainder residing within cells. The average daily Western diet contains 50–100 mEq of potassium, which is completely and passively absorbed in the upper gastrointestinal (GI) tract. To enter cells, potassium must first pass through the extracellular compartment. If the serum potassium concentrations rise above 6 mEq/L (>6 mmol/L), symptomatic hyperkalemia may occur. Potassium homeostasis is altered by insulin, aldosterone, changes in acid–base balance, renal function, or GI and skin losses. These conditions can be modulated by various pathological states as well as pharmacotherapy. Although potassium may affect different bodily functions, its effect on cardiac muscle is the most important due to the potential life-threatening effect of arrhythmias, as a result of either high or low serum potassium concentrations.4,6-10
Renal Homeostasis
When the serum potassium concentration is high, the body has two different mechanisms to restore potassium balance. One quick way is to shift the plasma potassium into cells, while the other slower mechanism is renal elimination.10 The kidneys are the primary organs involved in the control and elimination of potassium. Potassium is freely filtered at the glomeruli and almost completely reabsorbed before the filtrate reaches the collecting tubules. However, an amount equal to about 10% of the filtered potassium is secreted into the urine at the distal and collecting tubules. Virtually all the potassium recovered in urine is, therefore, delivered via tubular secretion rather than glomerular filtration.7
In the distal tubule, potassium is being secreted into the tubule while sodium is reabsorbed. There are several mechanisms that can modulate this sodium-potassium exchange. Aldosterone plays an important role since it increases potassium secretion into the urine (Figures 6-1 and 6-3).10 The hormone is secreted by the adrenal glands in response to high serum potassium concentrations. The delivery of large quantities of sodium and fluid to the distal tubules may also cause potassium secretion and its subsequent elimination, as seen in diuretic-induced hypokalemia.11 As the delivery of sodium and fluid is decreased, potassium secretion declines.
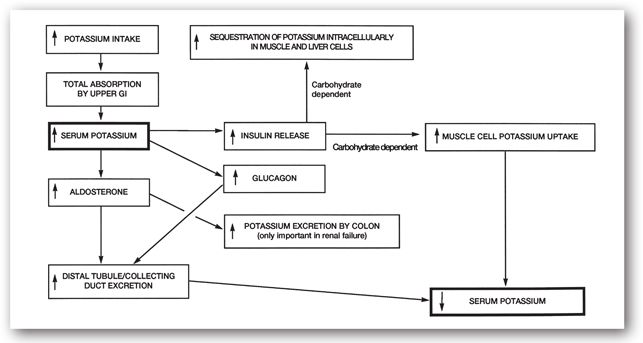
FIGURE 6-3. The acute homeostatic sequence of events in the body to maintain serum potassium within a narrow concentration range.
The presence of anions in the distal tubules, which are relatively permeable to reabsorption, can increase renal potassium loss because the negatively charged anions attract positively charged potassium ions. This mechanism is responsible for hypokalemia caused by renal tubular acidosis and the administration of high doses of drugs with sodium salts (e.g., penicillin and its derivatives).10 Potassium secretion is also influenced by the potassium concentration in distal tubular cells. When the intracellular potassium concentration is high, such as during dehydration, potassium secretion into the urine is increased.
In addition to the mechanisms addressed above, there are several additional points that we should consider regarding the maintenance of potassium homeostasis:
- Although the kidneys are the primary route of elimination, potassium secretion into the colon becomes important in patients with advanced renal failure.10
- Unlike sodium, the kidneys are not fully able to arrest potassium’s secretion into the urine. During hypokalemia, the urinary potassium concentration may decrease to as low as 5 mEq/L, however, potassium excretion does not cease completely.
The modulation of renal potassium excretion by these mechanisms may take hours to cause significant changes in serum potassium concentration, even during drastic, acute changes. Extrarenal mechanisms, therefore, often play important roles in keeping the serum potassium concentration within the narrow acceptable range.
Acid–Base Homeostasis
Another potentially relevant factor influencing renal potassium secretion is serum pH. When arterial pH increases due to metabolic, but not respiratory, alkalosis, a compensatory efflux of hydrogen ions from the cells into the extracellular fluid (bloodstream) takes place with a concurrent influx of potassium ions into the cells in order to maintain an electropotential gradient.7 During the early phase of metabolic alkalosis, the serum potassium concentration is reduced due to a pH-dependent intracellular influx of serum potassium from the serum without altering the total body amount. Thus, although there is no immediate change in the amount of total body potassium, this movement of ions increases the cellular potassium content and results in hypokalemia.
However, a shift in potassium and hydrogen ions also takes place in the renal distal tubular cells. In the presence of persistent alkalemia, renal potassium secretion into the urine is increased. Over time, the serum potassium concentration declines through increased renal loss, resulting in a reduced body store.
Metabolic acidosis has the opposite effect. Decreased pH results in an extracellular shift of potassium as a result of an intracellular shift of hydrogen ions, causing an elevated serum potassium concentration.7 Since the intracellular potassium content of the distal tubular cell is decreased, secretion of potassium in the urine is diminished. Chronically, however, renal potassium loss gradually increases due to unknown mechanisms.
When a severe metabolic acid–base abnormality exists, adjustment of the measured serum potassium concentration may be necessary to more accurately assess the body potassium status. For every 0.1-unit reduction in arterial pH less than 7.4, roughly 0.6 mEq/L (range: 0.2–1.7 mEq/L) could be added to the serum potassium value:
Kcorr = ([7.4 – pH]/0.1 × 0.6 mEq/L) + Kuncorr
where Kcorr is the corrected serum potassium concentration and Kuncorr is the uncorrected or measured serum potassium concentration (Minicase 3).7 It is important to note that Kcorr is a hypothetical value and only reflects what the serum potassium concentration would be if the serum pH is normalized and in the absence of other factors affecting potassium homeostasis. As long as the serum pH remains abnormal, the measured serum potassium concentration (Kuncorr) is the true reflection of actual serum potassium concentration. The Kcorr value should always be assessed together with the actual serum potassium concentration and the patient’s clinical presentation. The clinical value of calculating Kcorr is mostly to avoid overcorrection of potassium based solely on Kuncorr, as well as to provide a more complete picture that reflects total potassium stores in the body. Clinicians should remember that regardless of the value of Kcorr, a patient with a significantly abnormal measured (uncorrected) serum potassium concentration is still at risk for developing cardiac arrhythmias.
A Case of Hyperkalemia
JAMES D., A 65-YEAR-OLD, 150-LB MAN, was admitted to University Hospital for surgical management of acute urinary retention due to bladder outlet obstruction. His past medical history was significant for hypertension and diet-controlled type 2 diabetes mellitus. His medications (all orally) included atenolol 100 mg daily, hydrochlorothiazide 25 mg daily, and enalapril 10 mg every morning.
Vital signs upon presentation included a respiratory rate 25 breaths per minute, pulse 60 beats per minute with regular rhythm, and BP 160/95 mm Hg. Remarkable findings from physical examination included an audible S4 heart sound and decreased deep-tendon reflexes bilaterally. The only other abnormal clinical finding was an indurated, fixed prostate gland. Abnormal clinical laboratory tests included serum potassium 5.6 mEq/L (3.8–5.0 mEq/L), serum total carbon dioxide 16 mmol/L (24–30 mmol/L), glucose 237 mg/dL (70–110 mg/dL), phosphate 6.4 mg/dL (2.3–4.7 mg/dL), BUN 56 mg/dL (8–23 mg/dL), and SCr 3.3 mg/dL (0.6–1.2 mg/dL). A presurgery arterial blood gas showed pH 7.30 (7.36–7.44), partial pressure of arterial carbon dioxide (PaCO2) 17 mm Hg (35–40 mm Hg), and partial pressure of arterial oxygen (PaO2) 99 mm Hg (95–100 mm Hg) on room air.
Twenty-four hours after surgery, he complained of muscle weakness and developed anuria. His pulse was around 40 beats per minute and with irregular rhythms, and his BP was 130/70 mm Hg. His EKG showed flat P waves, tall T waves, and widened QRS intervals. Repeat serum chemistry test revealed serum potassium concentration of 7.1 mEq/L (3.8–5.0 mEq/L).
Question: What are the potential causes of symptomatic hyperkalemia in James D.?
Discussion: James D. is exhibiting signs and symptoms of hyperkalemia (e.g., muscle weakness, rhythm disturbances, worsening of bradycardia, and reduced BP). He has several predisposing factors that may have contributed to his hyperkalemia. Acute renal failure is the most significant cause. If his underlying metabolic acidosis has not yet been corrected, extracellular shifting of potassium could occur leading to an elevated serum potassium concentration. Although clinically insignificant, his serum potassium value may be corrected for his acidosis to more accurately reflect his extracellular potassium status. The corrected serum potassium value is
Kcorr = ([7.4 – pH]/0.1 × 0.6 mEq/L) + Kuncorr
Kcorr = ([7.4 – 7.3]/0.1 × 0.6 mEq/L) + 7.1 = 7.7 mEq/L
This correction implies that if the acidemia is treated to restore plasma pH to 7.4, the serum potassium concentration will increase by 0.6 mEq/L without any addition of potassium to the body. Such correction becomes more significant when the metabolic acid–base disorder is more severe.
Medications such as ACE-inhibitors can also contribute to hyperkalemia, although in this case, the impact is likely to be limited because (1) the dose used is small; (2) it is a chronic medication; and (3) the presence of acute renal failure would have a much more immediate and profound impact in elevating serum potassium.
Figure 6-3 summarizes the acute homeostatic mechanism involved in potassium distribution. During hyperkalemia, along with the release of aldosterone, increased glucagon and insulin release also contribute to reducing the serum potassium concentration. Glucagon stimulates potassium secretion into the distal tubules and collecting ducts, while insulin promotes intracellular potassium uptake. Although insulin is not a major controlling factor in potassium homeostasis, it is useful for the emergency treatment of hyperkalemia.7,12
Pharmacological stimulation of beta-2 adrenergic receptors may also affect the transcellular equilibrium of potassium. It leads to the movement of potassium from extracellular fluid to the intracellular fluid compartment. Beta-2 adrenergic agonists (e.g., albuterol) can, therefore, be used short term to treat certain hyperkalemic patients.7-9
Hypokalemia
Hypokalemia is defined as a serum potassium concentration less than 3.8 mEq/L (<3.8 mmol/L).10 To interpret the significance of low potassium values, clinicians should determine whether hypokalemia is due to intracellular shifting of potassium (apparent deficit) or increased loss from the body (true deficit) (List 6-4).
Intracellular shifting occurs as a result of metabolic alkalosis or after administration of insulin, or large doses of beta-2 adrenergic agonists (e.g., continuous or hourly use of albuterol in ICU patients receiving mechanical ventilation).9,13 Increased elimination of potassium can occur in the kidneys or GI tract. There may be decreased potassium reabsorption in the proximal tubules or increased secretion in the distal tubules and collecting ducts.10
Amphotericin B. Proximal tubular damage can occur with amphotericin B therapy, resulting in renal tubular acidosis. Impaired reabsorption of potassium, magnesium, and bicarbonate may lead to hypokalemia, hypomagnesemia, and metabolic acidosis.7,13 A concurrent deficiency in magnesium may affect the ability to restore potassium balance. Magnesium maintains the sodium–potassium adenosine triphosphate (ATP) pump activity and facilitates renal preservation of potassium. A hypokalemic patient who is also hypomagnesemic may not, therefore, respond to potassium replacement therapy unless the magnesium balance is restored.7,8,12 It appears that the lipid formulations of amphotericin B may still affect potassium homeostasis, although the magnitude may be less severe and the presentation is less acute.
Diuretics. Nonpotassium-sparing diuretic agents are drugs most commonly associated with renal potassium wasting. Although their mechanisms of natriuretic action differ, diuretic-induced hypokalemia is primarily caused by increased secretion of potassium at the distal sites in the nephron in response to an increased load of exchangeable sodium. Diuretics increase the distal urinary flow by inhibiting sodium reabsorption. This increased delivery of fluid and sodium in the distal segment of the nephron results in an increase in sodium reabsorption at that site. To maintain a neutral electropotential gradient in the lumen, potassium is excreted as sodium is reabsorbed. Therefore, any inhibition of sodium absorption by diuretics proximal to or at the distal tubules can increase potassium loss. Renal potassium excretion is further enhanced when nonabsorbable anions are present in the urine.
Use of loop diuretics (e.g., furosemide) and thiazides (e.g., hydrochlorothiazide) may both result in hypokalemia and the effect is dose-dependent. Serum potassium concentrations should be monitored regularly, especially in patients receiving high doses of loop diuretics, to avoid the increased risk of cardiovascular events secondary to hypokalemia and other electrolyte imbalances. In addition, elderly patients with ischemic heart disease and patients receiving digoxin are more susceptible to the adverse consequences of hypokalemia.15-17 Other drugs commonly used in managing hypertension and other cardiac diseases such as spironolactone, triamterene, amiloride, eplerenone, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor antagonists are not expected to cause potassium loss due to their mode of action. On the contrary, they cause retention of potassium due to their effects related to aldosterone-dependent exchange sites in the collecting tubules.11,18
Other causes. Conditions that cause hyperaldosteronism, either primary (e.g., adrenal tumor) or secondary (e.g., renovascular hypertension), can produce hypokalemia.13 Cushing syndrome leads to increased circulation of mineralocorticoids such as aldosterone. Corticosteroids with strong mineralocorticoid activity (e.g., cortisone) also can cause hypokalemia.10
GI loss of potassium can be important. Aldosterone influences both renal and intestinal potassium handling.10 A decrease in extracellular volume increases aldosterone secretion, resulting in renal and colonic potassium wasting. Diarrheal fluid can contain 20–120 mEq/L (20–120 mmol/L) of potassium; consequently, profuse diarrhea can rapidly result in potassium imbalance. In contrast, vomitus contains only 0–32 mEq/L (0–32 mmol/L) of potassium, and loss secondary to vomiting is unlikely to be significant. However, with severe vomiting, the resultant metabolic alkalosis may lead to hypokalemia due to intracellular shifting of potassium. Finally, patients receiving potassium-free parenteral fluids can become hypokalemic if not monitored properly.7,10
Clinical diagnosis. Signs and symptoms of hypokalemia involve many physiological systems. Abnormalities in the cardiovascular system may result in serious consequences (i.e., disturbances in cardiac rhythm). Hypokalemia-induced arrhythmias are of particular concern in patients receiving digoxin. Both digitalis glycosides and hypokalemia inhibit the sodium-potassium ATP pump in the cardiac cells. Together, they can deplete intracellular potassium, which may result in fatal arrhythmias. The signs and symptoms of hypokalemia are listed in List 6-5. Skeletal muscle weakness is often seen; severe depletion may lead to decreased reflexes and paralysis. Death can occur from respiratory muscle paralysis.7,9,19
Hyperkalemia
Hyperkalemia is defined as a serum potassium concentration greater than 5.0 mEq/L (>5.0 mmol/L). As with hypokalemia, hyperkalemia may indicate a true or apparent potassium imbalance, although the signs and symptoms are indistinguishable.10 To interpret a high serum potassium value, the clinician should determine whether hyperkalemia is due to apparent excess caused by extracellular shifting of potassium or true potassium excess in the body caused by increased intake with diminished excretion (List 6-6).4,6,9,11
Causes. Since renal excretion is the major route of potassium elimination, renal failure is the most common cause of hyperkalemia. However, potassium handling by the nephrons is relatively well-preserved until the GFR falls to less than 10% of normal. Many patients with renal impairment can, therefore, maintain a near normal, serum potassium concentration. They are still prone to developing hyperkalemia if excessive potassium is consumed and when renal function deteriorates.9,12
Increased potassium intake rarely causes any problem in subjects in the absence of significant renal impairment. With normal renal function, increased potassium intake will lead to increased renal excretion and redistribution to the intracellular space through the action of aldosterone and insulin, respectively. Interference with either mechanism may result in hyperkalemia. Decreased aldosterone secretion can occur with Addison disease or other defects affecting the hormone’s adrenal output.7,12 Pathological changes affecting the proximal or distal renal tubules can also lead to hyperkalemia.7,12
Use of potassium-sparing diuretics (e.g., spironolactone) is a common cause of hyperkalemia, especially in patients with renal function impairment. Concurrent use of potassium supplements (including potassium-rich salt substitutes) will also increase the risk.
Similar to hypokalemia, hyperkalemia can result from transcellular shifting of potassium. In the presence of severe acidemia, potassium shifts from the intracellular to the extracellular space, which may result in a clinically significant increase in the serum potassium concentration.10
Clinical diagnosis. The cardiovascular manifestations of hyperkalemia are of major concern. They include cardiac rhythm disturbances, bradycardia, hypotension, and, in severe cases, cardiac arrest. At times, muscle weakness may occur before these cardiac signs and symptoms. To appreciate the potent effect of potassium on the heart, one has to realize that potassium is the principal component of cardioplegic solutions commonly used to arrest the rhythm of the heart during cardiac surgeries.7,9,12
Causes of spurious laboratory results. There are several conditions that will result in fictitious hyperkalemia in which the high serum concentration reported is not expected to have any significant clinical sequelae. Erythrocytes, similar to other cells, have high potassium content. When there is substantial hemolysis in the specimen collection tube, the red cells will release potassium in quantities large enough to produce misleading results. Hemolysis may occur when a very small needle is used for blood draw, the tourniquet is too tight, or when the specimen stands too long or is mishandled. When a high serum potassium concentration is reported in a patient without pertinent signs and symptoms, the test needs to be repeated to rule out hemolysis.6,7,10
A similar phenomenon can occur when the specimen is allowed to clot (when nonheparinized tubes are used) because platelets and white cells are also rich in potassium. In patients with leukemia or thrombocytosis, the potassium concentration should be obtained from plasma rather than serum samples. However, the normal plasma potassium concentration is 0.3–0.4 mEq/L lower than the serum values.
Chloride
Normal range: 95–103 mEq/L or 95–103 mmol/L
Physiology
Chloride is the most abundant extracellular anion. However, its intracellular concentration is small (about 4 mEq/L). Chloride is passively absorbed from the upper small intestine. In the distal ileum and large intestine, its absorption is coupled with bicarbonate ion secretion. Chloride is primarily regulated by the renal proximal tubules, where it is exchanged for bicarbonate ions. Throughout the rest of the nephron, chloride passively follows sodium and water.
Chloride is influenced by the extracellular fluid balance and acid–base balance.19,20 Although homeostatic mechanisms do not directly regulate chloride, they indirectly regulate it through changes in sodium and bicarbonate. The physiological role of chloride is primarily passive. It balances out positive charges in the extracellular fluid and, by passively following sodium, helps to maintain extracellular osmolality.
Hypochloremia and Hyperchloremia
Serum chloride values are used as confirmatory tests to identify fluid balance and acid–base abnormalities.21 Like sodium, a change in the serum chloride concentration does not necessarily reflect a change in total body content. Rather, it indicates an alteration in fluid status and/or acid–base balance. One of the most common causes of hyperchloremia in hospitalized patients results from saline infusion. Chloride has the added feature of being influenced by bicarbonate. Therefore, it would be expected to decrease to the same proportion as sodium when serum is diluted with fluid and to increase to the same proportion as sodium during dehydration. However, when a patient is on acid-suppressive therapy (e.g., high-dose H2-blockers or proton pump inhibitors), has been receiving continuous or frequent nasogastric suction, or has profuse vomiting, a greater loss of chloride than sodium can occur because gastric fluid contains 1.5–3 times more chloride than sodium. Gastric outlet obstruction, protracted vomiting and self-induced vomiting can also lead to hypochloremia.
Drug and parenteral nutrition causes. Even though drugs can influence serum chloride concentrations, they rarely do so directly. For example, although loop diuretics (e.g., furosemide) and thiazide diuretics (e.g., hydrochlorothiazide) inhibit chloride uptake at the loop of Henle and distal nephron, respectively, the hypochloremia that may result is due to the concurrent loss of sodium and contraction alkalosis.18,21 Since chloride passively follows sodium, salt and water retention can transiently raise serum chloride concentrations. This effect occurs with corticosteroids, guanethidine, and nonsteroidal anti-inflammatory agents (NSAIDs) such as ibuprofen. Also, parenteral nutrition solutions with high chloride concentrations are associated with an increased risk of hyperchloremia. Acetate or phosphate salts used in place of chloride salts (e.g., potassium chloride) reduce this risk. Acetazolamide also can cause hyperchloremia.
Acid–base status and other causes. The acid–base balance is partly regulated by renal production and excretion of bicarbonate ions. The proximal tubules are the primary regulators of bicarbonate. These cells exchange bicarbonate with chloride to maintain the intracellular electropotential gradient. Renal excretion of chloride increases during metabolic alkalosis, resulting in a reduced serum chloride concentration.
The opposite situation also may be true: metabolic or respiratory acidosis results in an elevated serum chloride concentration. Hyperchloremic metabolic acidosis is not common but may occur when the kidneys are unable to conserve bicarbonate, as in interstitial renal disease (e.g., obstruction, pyelonephritis, and analgesic nephropathy), GI bicarbonate loss (e.g., cholera and staphylococcal infections of the intestines), and acetazolamide-induced carbonic anhydrase inhibition. Falsely elevated chloride rarely occurs from bromide toxicity due to a lack of distinction between these two halogens by the laboratory’s chemical analyzer.
Since the signs and symptoms associated with hyperchloremia and hypochloremia are related to fluid status or the acid–base balance and underlying causes, rather than to chloride itself, the reader is referred to discussions in Chapter 9: Arterial Blood Gases and Acid–Base Balance.
OTHER MINERALS
Magnesium
Normal range: 1.3–2.1 mEq/L or 0.65–1.05 mmol/L
Physiology
Magnesium has a widespread physiological role in maintaining neuromuscular functions and enzymatic functions. Magnesium acts as a cofactor for phosphorylation of ATPs from adenosine phosphates (ADPs). Magnesium is also vital for binding macromolecules to organelles (e.g., messenger ribonucleic acid [mRNA] to ribosomes).
The average adult body contains 21–28 g (1750–2400 mEq) of magnesium with the following distribution:
- About 50% in bone (about 30% or less of this pool is slowly exchangeable with extracellular fluid)
- 20% in muscle
- Around 10% in nonmuscle soft tissues
- 1% to 2% in extracellular fluid (for plasma magnesium, about 50% is free; approximately 15% is complexed to anions; and 30% is bound to protein, primarily albumin)
The average daily magnesium intake is 20–40 mEq/day. Approximately 30% to 40% of the ingested magnesium is absorbed from the jejunum and ileum through intercellular and intracellular pathways. Both passive diffusion down an electrochemical gradient and active transport process are involved. The extent of magnesium absorption may be affected by dietary magnesium intake, calcium intake, vitamin D, and PTH. However, conflicting data are available and the extent of these parameters in affecting absorption is unresolved. Certain medications (e.g., cyclosporine, tacrolimus, cisplatin, amphotericin B) can significantly increase renal magnesium loss, predisposing the patient to hypomagnesemia.
Urinary magnesium accounts for one-third of the total daily magnesium output while the other two-thirds are in the GI tract (e.g., stool). Magnesium is excreted in the kidneys, where unbound serum magnesium is freely filtered at the glomerulus. All but about 3% to 5% of filtered magnesium is normally reabsorbed (100 mg/day). In other words, 97% of the filtered magnesium is reabsorbed under normal circumstances. Reabsorption is primarily through the ascending limb of the loop of Henle (50% to 60%). About 30% is reabsorbed in the proximal tubule and 7% from the distal tubule. This explains why loop diuretics have a more profound effect on renal magnesium wasting. The drive of magnesium reabsorption is mediated by the charge difference generated by the sodium-potassium-chloride cotransport system in the lumen.
The regulation of magnesium is primarily driven by the plasma magnesium concentration. Changes in plasma magnesium concentrations have potent effects on renal reabsorption and stool losses. These effects are seen over 3–5 days and may persist for a long time. Hormonal regulation of magnesium seems to be much less critical for its homeostasis.
Factors affecting calcium homeostasis also affect magnesium homeostasis.22 A decline in serum magnesium concentration stimulates the release of PTH, which increases serum magnesium by increasing its release from the bone store and renal reabsorption. Hyperaldosteronism causes increased magnesium renal excretion. Insulin by itself does not alter the serum magnesium concentration. But in a hyperglycemic state, insulin causes a rapid intracellular uptake of glucose. This process causes an increase in the phosphorylation by sodium–potassium ATPase on the cell membrane. Since magnesium is a cofactor for sodium potassium ATPase, magnesium is consumed, which subsequently decreases the serum magnesium concentration. Excretion of magnesium is influenced by serum calcium and phosphate concentrations. Magnesium movement generally follows that of phosphate (i.e., if phosphate declines, magnesium also declines) and is the opposite of calcium.21,22 Other factors that increase magnesium reabsorption include acute metabolic acidosis, hyperthyroidism, and chronic alcohol use.
Hypomagnesemia
Hypomagnesemia is defined as a serum magnesium concentration less than 1.3 mEq/L (<0.65 mmol/L). The common causes of hypomagnesemia include renal wasting, chronic alcohol use, diabetes mellitus, protein-calorie malnutrition, refeeding syndrome, and postparathyroidectomy. Since serum magnesium deficiency can be offset by magnesium release from bone, muscle, and the heart, the serum value may not be a useful indicator of cellular depletion and complications (e.g., arrhythmias). However, low serum magnesium usually indicates low cellular magnesium as long as the patient has a normal extracellular fluid volume.22,23
Causes. Magnesium deficiency is more common than magnesium excess. Depletion usually results from excessive loss from the GI tract or kidneys (e.g., use of loop diuretics). Magnesium depletion is not commonly the result of decreased intake because the kidneys can cease magnesium elimination in 4–7 days to conserve the ion. However, with chronic alcohol consumption, deficiency can occur from a combination of poor intake, poor GI absorption (e.g., vomiting or diarrhea), and increased renal elimination. Depletion can also occur from poor intestinal absorption (e.g., small-bowel resection). Diarrhea can be a source of magnesium loss because diarrhea stools may contain as much as 14 mEq/L (7 mmol/L) of magnesium.
Urinary magnesium loss may result from diuresis or tubular defects, such as the diuretic phase of acute tubular necrosis. Some patients with hypoparathyroidism may exhibit low magnesium serum concentrations from renal loss and, possibly, decreased intestinal absorption. Other conditions associated with magnesium deficiency include hyperthyroidism, primary aldosteronism, diabetic ketoacidosis, and pancreatitis. Magnesium deficiency associated with these conditions may be particularly dangerous because often there are concurrent potassium and calcium deficiencies. Although loop diuretics lead to significant magnesium depletion, thiazide diuretics do not cause hypomagnesemia, especially at lower doses (50 mg/day). Furthermore, potassium-sparing diuretics (e.g., spironolactone, triamterene, and amiloride), are also magnesium-sparing and have some limited clinical role in diuretic-induced hypokalemia and hypomagnesemia.22,24
Clinical diagnosis. Magnesium depletion is usually associated with neuromuscular symptoms such as weakness, muscle fasciculation with tremor, tetany, and increased reflexes.22 They occur because the release of acetylcholine to motor endplates is affected by the presence or absence of magnesium. Motor endplate sensitivity to acetylcholine is also affected. When serum magnesium decreases, acetylcholine release increases, resulting in increased muscle excitation.
Magnesium also affects the central nervous system (CNS). Magnesium depletion can cause personality changes, disorientation, convulsions, psychosis, stupor, and coma.22,25 Severe hypomagnesemia may result in hypocalcemia due to intracellular cationic shifts. Many symptoms of magnesium deficiency result from concurrent hypocalcemia.
Perhaps the most important effects of magnesium imbalance are on the heart. Decreased magnesium in cardiac cells may manifest as a prolonged QT interval (increased risk of arrhythmias, especially torsades de pointes).25 Moderately decreased concentrations can cause electrocardiogram (EKG) abnormalities similar to those observed with hypokalemia. In addition, vasodilation may occur by a direct effect on blood vessels and ganglionic blockade.
A 24-hour urine magnesium excretion test may be helpful in determining the magnitude of a total body magnesium deficiency. If the value is normal, the patient is not considered deficient as long as serum magnesium is also normal. The diagnosis of total body magnesium deficiency is established when the 24-hour urinary magnesium excretion is low even in the presence of normal serum magnesium concentration.
Hypermagnesemia
Hypermagnesemia is defined as a serum magnesium concentration greater than 2.1 mEq/L (>1.05 mmol/L).
Causes. Besides magnesium overload (e.g., over-replacement of magnesium, treatment for preeclampsia, and antacid/laxative overuse), the most important risk factor for hypermagnesemia is renal dysfunction. Rapid infusions of IV solutions containing large amounts of magnesium, such as those given for myocardial infarction, preeclampsia, and status asthmaticus, may result in hypermagnesemia.
Clinical diagnosis. Plasma magnesium concentrations below 5 mEq/L (<2.5 mmol/L) rarely cause serious symptoms. No specific symptoms, such as muscle weakness, decrease in deep tendon reflexes or fatigue, may be present. As magnesium concentration rises above 5 mEq/L, more notable symptoms such as lethargy, mental confusion, and hypotension may be observed (Table 6-3).22,24,27 In severe hypermagnesemia (>10 mEq/L), life-threatening symptoms, including coma, paralysis, or cardiac arrest, can be observed and urgent therapy is indicated.
Treatment for severe or symptomatic hypermagnesemia may include IV calcium gluconate 1–2 g over 30 minutes to reverse the neuromuscular and cardiovascular blockade of magnesium. Increased renal elimination of magnesium can be achieved by forced diuresis with IV saline hydration and a loop diuretic agent. Hemodialysis should be reserved as a last resort.
Calcium
Normal range: 9.2–11.0 mg/dL or 2.3–2.8 mmol/L for adults
Physiology
Calcium plays an important role in the propagation of neuromuscular activity; regulation of endocrine functions (e.g., pancreatic insulin release and gastric hydrogen secretion), blood coagulation including platelet aggregation, and bone and tooth metabolism.2,28
The serum calcium concentration is closely regulated by complex interaction among PTH, serum phosphate, vitamin D system, and the target organ (Figure 6-4). About one-third of the ingested calcium is actively absorbed from the proximal area of the small intestine, facilitated by 1,25-dihydroxycholecalciferol (1,25-DHCC or calcitriol, the most active form of vitamin D). Passive intestinal absorption is negligible with intake of less than 2 g/day. The average daily calcium intake is 2–2.5 g/day.
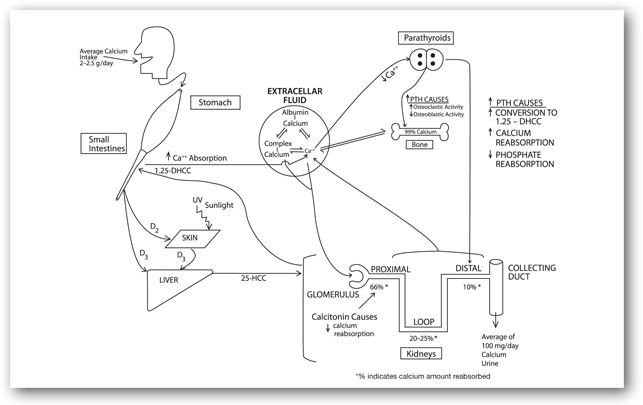
FIGURE 6-4. Calcium physiology: relationship with vitamin D, calcitonin, PTH, and albumin.
The normal adult body contains about 1000 g of calcium, with only 0.5% found in the extracellular fluid; 99.5% is integrated into bones. Therefore, the tissue concentration of calcium is small. Since bone is constantly remodeled by osteoblasts and osteoclasts, a small quantity of bone calcium is in equilibrium with the extracellular fluid. Extracellular calcium exists in three forms:
- Complexed to bicarbonate, citrates, and phosphates (6%)
- Protein bound, mostly to albumin (40%)
- Ionized or free fraction (54%)
Intracellular calcium. Imbalance of body calcium results in disturbances in muscle contraction and nerve action.28 Within the cells, calcium maintains a low concentration. The calcium that is attracted into the negatively charged cell is either actively pumped out or sequestered by mitochondria or the endoplasmic reticulum. Such differences in concentrations allow calcium to be used for transmembrane signaling. In response to stimuli, calcium is allowed either to enter a cell or released from internal cellular stores where it interacts with specific intracellular proteins to regulate cellular functions or metabolic processes.2,26,27 Calcium enters cells through one of the three types of calcium channels that have been identified: T (transient or fast), N (neuronal), and L (long lasting or slow). Subsets of these channels may exist. Calcium channel-blockers are likely to affect the L channels.29
In muscle, calcium is released from the intracellular sarcoplasmic reticulum. The released calcium binds to troponin and stops troponin from inhibiting the interaction of actin and myosin. This interaction results in muscle contraction. Muscle relaxation occurs when calcium is pumped back into the sarcoplasmic reticulum. In cardiac tissue, calcium becomes important during phase 2 of the action potential. During this phase, fast entry of sodium stops and calcium entry through the slow channels begins (Figure 6-5), resulting in contraction. During repolarization, calcium is actively pumped out of the cell.2
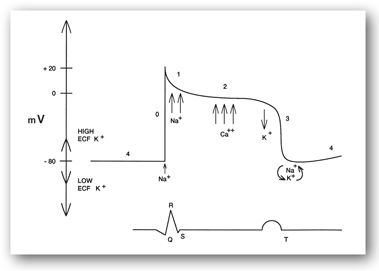
FIGURE 6-5. Cardiac intracellular potential and its relationship to the ECG.
Calcium channel-blocking drugs (e.g., nifedipine, diltiazem, and verapamil) inhibit the movement of calcium into muscle cells, thus decreasing the strength of contraction. The areas most sensitive to these effects appear to be the sinoatrial and atrioventricular nodes and vascular smooth muscles, which explains the hypotensive effects of nifedipine.
Extracellular calcium. Complex-bound calcium usually accounts for less than 1 mg/dL (<0.25 mmol/L) of blood calcium. The complex usually is formed with bicarbonate, citrate, or phosphate. In patients with CKD, calcium may also be bound with sulfate because the anion is retained. Phosphate plays an important role in calcium homeostasis. Under normal physiological conditions, the product of calcium concentration times phosphate concentration (the so-called calcium–phosphate product) is relatively constant: an increase in one ion necessitates a corresponding decline in the other. In addition, many homeostatic mechanisms that control calcium also regulate phosphate. This relationship is particularly important in renal failure; the decreased phosphate excretion may ultimately lead, through a complex mechanism, to hypocalcemia, especially if the hyperphosphatemia is untreated.30,31
Calcium is bound primarily to serum albumin (80%) and globulins (20%). Protein-bound calcium is in equilibrium with ionized calcium, which is affected by the serum anion concentration and blood pH. This equilibrium is important since ionized calcium is the physiologically active moiety. Alkalosis increases protein binding of calcium, resulting in a lower free fraction, whereas acidosis has the opposite effect. In patients with respiratory or metabolic alkalosis, the signs and symptoms of hypocalcemia may become more pronounced due to increased binding. Conversely, signs and symptoms of hypercalcemia become more apparent in patients with metabolic or respiratory acidosis. Therefore, total serum calcium concentration, which is commonly reported by clinical laboratories, is not as clinically significant as the quantity of available ionized calcium. In fact, it is the free calcium concentration that is closely regulated by the different homeostatic mechanisms.
Clinically, serum protein concentrations, especially albumin, have an important influence on regulating the amount of physiologically active calcium in the serum. The normal serum calcium range is 9.2–11.0 mg/dL (2.3–2.8 mmol/L) for a patient with a serum albumin of approximately 4 g/dL. In normal healthy adults, only 40% to 50% of the total serum calcium is free from protein-binding and thus considered as physiologically active. In patients with hypoalbuminemia (due to acute illnesses, severe malnutrition), the free concentration of calcium is elevated despite a “normal” total serum calcium concentration. Therefore, it is a common practice to either measure ionized calcium or to correct the total serum calcium concentration based on the measured albumin concentration. The following formula is commonly used in an attempt to “correct” total serum calcium concentration:
Cacorr = ([4.0 – albumin] × 0.8 mg/dL) + Cauncorr
where Cacorr is the corrected serum calcium concentration, and Cauncorr is the uncorrected (or measured total) serum calcium concentration. For example, a clinician may be asked to write parenteral nutrition orders for an emaciated cancer patient. The serum albumin is 1.9 g/dL (19 g/L), and the total serum calcium concentration is 7.7 mg/dL (1.9 mmol/L). At first glance, one might consider the calcium to be low. But with the reduced serum albumin concentration, more ionized calcium is available to cells.
Cacorr = ([4.0 – 1.9] × 0.8) + 7.7 = 9.4 mg/dL (2.34 mmol/L)
The corrected serum calcium concentration is, thus, within the normal range. More importantly, the patient does not exhibit any signs and symptoms of hypocalcemia. Calcium supplementation is not therefore indicated. In the presence of severe hypoalbuminemia, as in nutritionally deprived patients, an apparently low total serum calcium may in fact be sufficient or in some instances, excessive. If such a patient were given IV albumin, the free concentration of calcium would acutely decline due to the resultant increased binding. The measured total calcium concentration will need to be corrected with the new albumin concentration.
Although this serum calcium correction method may be useful, the clinician must be aware of its limitations and potential for inaccuracy. The correction factor of 0.8 represents an average fraction of calcium bound to albumin under normal physiology. To have an accurate determination of the free concentration, a direct measurement of serum ionized calcium concentration should be available in most clinical laboratories (normal range: 4.0–4.8 mg/dL or 1.00–1.20 mmol/L). Ultimately, the patient’s clinical presentation is the most important factor to determine if immediate treatment for a calcium disorder is indicated.
Although calcium absorption takes place throughout the entire small intestine, the proximal region of the small intestine ( jejunum and proximal ileum) are the most active and regulated areas. Calcium absorption from the human GI tract is mediated by two processes: (1) transcellular active transport, a saturable, vitamin D–responsive process mediated by specific calcium binding proteins primarily in the upper GI tract, particularly in the distal duodenum and upper jejunum; and (2) paracellular processes, a nonsaturable linear transfer via diffusion that occurs throughout the entire length of the intestine. Under normal physiology, the total calcium absorptive capacity is the highest in the ileum because of the longer residence time. The rate of paracellular calcium absorption is fairly stable regardless of calcium intake. However, when dietary calcium intake is relatively limited, the efficiency of transcellular calcium transport becomes higher and accounts for a significant fraction of the absorbed calcium. Transcellular calcium transport is closely regulated by vitamin D, although other mechanisms may also be involved. Specifically, 1,25-DHCC induces the intestinal expressions of transcellular calcium transporters through its binding with the vitamin D receptors (VDR) in the intestinal epithelial cells.
Effect of vitamin D. A small amount of calcium is excreted daily into the GI tract through saliva, bile, and pancreatic and intestinal secretions. However, the primary route of elimination is filtration by the kidneys. Calcium is freely filtered at the glomeruli, where approximately 65% is reabsorbed at the proximal tubules under partial control by calcitonin and 1,25-DHCC. Roughly 25% is reabsorbed in the loop of Henle, and another 10% is reabsorbed at the distal tubules under the influence of PTH.
Despite being classified as a vitamin, the physiological functions of vitamin D more closely resemble a hormone. Vitamin D is important for
- Intestinal absorption of calcium
- PTH-induced mobilization of calcium from bone
- Calcium reabsorption in the proximal renal tubules
Vitamin D must undergo several conversion steps before the active form, calcitriol or 1,25-DHCC, is formed. It is absorbed by the intestines in two forms, 7-dehydrocholesterol and cholecalciferol (vitamin D3). 7-dehydrocholesterol is converted into cholecalciferol in the skin by the sun’s ultraviolet radiation. Rickets, one of the causes of childhood hypocalcemia, is caused by reduced exposure to sunlight, resulting in diminished conversion of 7-dehydrocholesterol to cholecalciferol.
Hepatic and intestinal enzymes, including CYP27A1, CYP2J2 and CYP3A4, convert cholecalciferol to 25-hydroxycholecalciferol (25-HCC), which is then further activated by CYP27B1 in the kidneys to form the active 1,25-DHCC. This last conversion step is regulated by PTH. When PTH is increased during hypocalcemia, renal production of 1,25-DHCC increases, which increases intestinal absorption of calcium. 1,25-DHCC may in turn regulate PTH synthesis and secretion.30,32
Influence of calcitonin. Calcitonin is a hormone secreted by specialized C cells of the thyroid gland in response to a high level of circulating ionized calcium. Calcitonin inhibits osteoclastic activity, thereby inhibiting bone resorption. It also decreases calcium reabsorption in the renal proximal tubules to result in increased renal calcium clearance.28 Calcitonin is used for the treatment of acute hypercalcemia and several different forms of the hormone are available.
Influence of parathyroid hormone. Parathyroid hormone is the most important hormone involved in calcium homeostasis. It is secreted by the parathyroid glands, which are embedded in the thyroid, in direct response to low circulating ionized calcium. Parathyroid hormone closely regulates, and is also regulated by, the vitamin D system to maintain the serum ionized calcium concentration within a narrow range. Generally, PTH increases the serum calcium concentration and stimulates the enzymatic activity of CYP27B1 to promote renal conversion of 25-HCC to 1,25-DHCC, which enhances intestinal calcium absorption. Conversely, 1,25-DHCC is a potent suppressor of PTH synthesis via a direct mechanism that is independent of the serum calcium concentration.28,31 The normal reference range for serum PTH concentrations is 10–65 pg/mL.
Tubular reabsorption of calcium and phosphate at the distal nephron is controlled by PTH; it increases renal reabsorption of calcium and decreases the reabsorption of phosphate, resulting in lower serum phosphate and higher serum calcium concentrations. Perhaps the most important effect of PTH is on the bone. In the presence of PTH, osteoblastic activity is diminished and bone-resorption processes of osteoclasts are increased. These effects increase serum ionized calcium, which feeds back to the parathyroid glands to decrease PTH output.30
The suppressive effect of 1,25-DHCC (calcitriol) on PTH secretion is used clinically in patients with CKD who have excessively high serum PTH concentrations due to secondary hyperparathyroidism. Parathyroid hormone is a known uremic toxin, and its presence in supraphysiological concentrations has many adverse effects (e.g., suppression of bone marrow erythropoiesis and increased osteoclastic bone resorption with replacement by fibrous tissue).33 Figure 6-6 depicts the relationship between serum PTH and serum calcium concentrations.
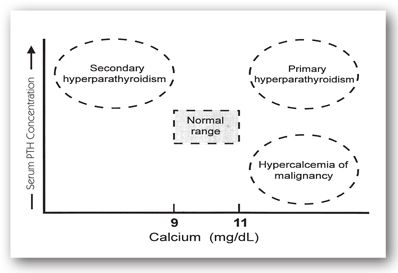
FIGURE 6-6. Interpretation of serum PTH concentrations with concomitant serum calcium concentrations.
Abnormalities. True abnormal serum concentrations of calcium may result from an abnormality in any of the previously mentioned mechanisms, including
- Altered intestinal absorption8,30,31,34
- Altered number or activity of osteoclast and osteoblast cells in bone8,30,31,34
- Changes in renal reabsorption of calcium8,30,31,34
- Calcium or phosphate IV infusions
Patients with CKD have increased serum phosphate and decreased serum calcium concentrations as a result of the following factors that interact via a complex mechanism: decreased phosphate clearance by the kidneys, decreased renal production of 1,25-DHCC, and skeletal resistance to the calcemic action of PTH. This interaction is further complicated by the metabolic acidosis of renal failure, which can increase bone resorption to result in decreased bone integrity.
Hypocalcemia
Hypocalcemia indicates a total serum calcium concentration of less than 9.2 mg/dL (<2.3 mmol/L). The most common cause of hypocalcemia is low serum proteins. As discussed previously, decreased serum protein leads to an increased free fraction of ionized calcium. If there is no other coexisting factor that could impair or alter calcium homeostasis, this should not be associated with a functional calcium deficit and clinical symptoms. Therefore, serum protein concentration should always be taken into consideration when interpreting serum total calcium concentration. Even in the case of true, mild hypocalcemia, the patient may remain asymptomatic and often no treatment is required.
The most common causes of a true reduction in total serum calcium are disorders of vitamin D metabolism or impaired PTH production (List 6-7). Osteomalacia (in adults) and rickets (in children) can result from severe deficiency in dietary calcium or vitamin D, diminished synthesis of vitamin D3 from insufficient sunlight exposure, or resistance of the intestinal wall to the action of vitamin D. The reduction in serum calcium leads to secondary hyperparathyroidism, which increases bone resorption. Over a long period to time, bones lose their structural integrity and become more susceptible to fracture. The diminished serum calcium concentration, if significant, may result in tetany.
Diminished intake. Although uncommon, diminished intake of calcium is an important cause of hypocalcemia, especially in patients receiving long-term total parental nutrition solutions.37,38
Medications. Excessive use of certain drugs to lower serum calcium by either increasing bone deposition or decreasing renal reabsorption of calcium may lead to hypocalcemia. These drugs include plicamycin, calcitonin, glucocorticoids, loop diuretics, etidronate, pamidronate, and alendronate.
Intravenous bicarbonate administration and hyperventilation can cause systemic alkalosis, resulting in decreased ionized serum calcium. This decrease is usually important only in patients who already have low serum calcium concentrations. Other drugs that may cause hypocalcemia include phenytoin, phenobarbital, aluminum-containing antacids, cisplatin, theophylline, sodium fluoride, and magnesium sulfate.
Another cause is rapid IV administration of phosphate salts, especially at high doses. Phosphate can bind calcium and form an insoluble complex that can deposit into soft tissues and clog the microcirculation, causing metastatic calcification, hardening of normally pliable tissues, or blockage of capillary blood flow.37,38 Soft-tissue deposition of the calcium–phosphate complex in lungs and blood vessels occurs when the serum solubility product of calcium times phosphate is high. The product of serum calcium and phosphate concentrations (both expressed in mg/dL) is often calculated, especially in patients with CKD, to minimize the risk for tissue calcification. The risk of deposition is higher in patients with a calcium–phosphate product that exceeds 50 or in patients with alkalosis. Other than the IV route, a large amount of phosphate may be absorbed from the GI tract with the use of certain enema and laxative preparations (e.g., Fleet’s enema or Fleet’s Phospho-Soda).33
Hypoparathyroidism. Hypoparathyroidism can reduce serum calcium concentrations. The most common cause of hypoparathyroidism is thyroidectomy, when the parathyroid glands are removed along with the thyroid glands. Since PTH is the major hormone regulating calcium balance, its absence significantly reduces serum calcium.38
Hyperparathyroidism. Hypocalcemia is commonly seen in patients with secondary hyperparathyroidism resulting from CKD (see Figure 6-6). The mechanism is complex and involves elevated serum phosphate concentrations and reduced activation of vitamin D. Parathyroid hormone acts on bone to increase calcium and phosphate resorption. Since renal phosphate elimination is reduced because of renal failure, the serum phosphate concentration is often high and depresses the serum calcium level. Because of the high phosphate concentrations in the intestinal lumen, dietary calcium is bound and absorption is impaired while phosphate absorption continues.
Metabolic acidosis. Common in CKD, metabolic acidosis further enhances bone resorption. With prolonged severe hyperparathyroidism, excessive osteoclastic resorption of bones results in replacement of bone material with fibrous tissues. This condition is termed osteitis fibrosa cystica.31,34,35 Such diminution of bone density may result in pathological fractures. Although the serum total calcium concentrations are low, patients may not show symptoms of hypocalcemia because the accompanying acidosis helps to maintain ionized serum calcium through the reduction in protein binding.
Magnesium. Similar to potassium, calcium balance depends strongly on magnesium homeostasis. Therefore, if a hypocalcemic patient is also hypomagnesemic, as a result of loop diuretic therapy, calcium replacement therapy may not be effective unless magnesium balance is restored.
Clinical diagnosis. As with any electrolyte disorder, the severity of the clinical manifestations of hypocalcemia depends on the acuteness of onset. Hypocalcemia can at times be a medical emergency with symptoms primarily in the neuromuscular system.37,38 They include fatigue, depression, memory loss, hallucinations, and, in severe cases, seizures, and tetany. The early signs of hypocalcemia are finger numbness, tingling and burning of extremities, and paresthesia. Mental instability and confusion may be seen in some patients as the primary manifestation.
Tetany is the hallmark of severe hypocalcemia. The mechanism of muscle fasciculation during tetany is the loss of the inhibitory effect of ionized calcium on muscle proteins. In extreme cases, this loss leads to increased neuromuscular excitability that can progress to laryngospasm and tonic-clonic seizures. Chvostek and Trousseau signs are hallmarks of hypocalcemia. Chvostek sign is a unilateral spasm induced by a slight tap over the facial nerve. Trousseau sign is a carpal spasm elicited when the upper arm is compressed by a blood pressure cuff.31,38,40
As hypocalcemia worsens, the cardiovascular system may be affected, as evidenced by myocardial failure, cardiac arrhythmias, and hypotension.37,38 Special attention should be given to serum calcium concentrations in patients receiving diuretics, corticosteroids, digoxin, antacids, lithium, and parenteral nutrition and in patients with renal disease.
Hypercalcemia
Hypercalcemia indicates a total serum calcium concentration greater than 11.0 mg/dL (>2.8 mmol/L).
Causes. The most common causes of hypercalcemia are malignancy and primary hyperparathyroidism (see Figure 6-6). Malignancies can increase serum calcium by several mechanisms. Osteolytic metastases can arise from breast, lung, thyroid, kidney, or bladder cancer. These tumor cells invade bone and produce substances that directly dissolve bone matrix and mineral content. Some malignancies, such as multiple myeloma, can produce factors that stimulate osteoclast proliferation and activity. Another mechanism is the ectopic production of PTH or PTH-like substances by tumor cells, resulting in a pseudohyperparathyroid state.39,41
In primary hyperparathyroidism, inappropriate secretion of PTH from the parathyroid gland, usually due to adenoma, increases serum calcium concentrations. The other major cause of hypercalcemia in hyperparathyroidism is the increased renal conversion of 25-HCC to active 1,25-DHCC. As the serum calcium concentration rises, the renal ability to reabsorb calcium may be exceeded, leading to an increased urinary calcium concentration and the subsequent formation of calcium–phosphate and calcium–oxalate renal stones. Typically, this condition results from parathyroid adenomas but may also be caused by primary parathyroid hyperplasia of chief cells or parathyroid carcinomas.31,41
Approximately 2% of patients treated with thiazide diuretics may develop hypercalcemia. Patients at risk are those with hyperparathyroidism. The mechanism appears to be multifactorial and includes enhanced renal reabsorption of calcium and decreased plasma volume.
The milk-alkali syndrome (Burnett syndrome), rarely observed today, is another drug-related cause of hypercalcemia.33 This syndrome occurs from a chronic high intake of milk or calcium products combined with an absorbable antacid (e.g., calcium carbonate, sodium bicarbonate, or magnesium hydroxide). This syndrome was more common in the past when milk or cream was used to treat gastric ulcers and before the advent of nonabsorbable antacids. Renal failure can occur as a result of calcium deposition in soft tissues.33,42
Hypercalcemia can also result from the following28,40,41,43:
- Excessive administration of IV calcium salts
- Calcium supplements
- Chronic immobilization
- Paget disease
- Sarcoidosis
- Hyperthyroidism
- Acute adrenal insufficiency
- Some respiratory diseases
- Lithium-induced renal calcium reabsorption
- Excessive vitamin D, vitamin A, or thyroid hormone, which increases intestinal absorption
- Tamoxifen
- Androgenic hormones
- Estrogen
- Progesterone
Clinical diagnosis. Similar to hypocalcemia and other electrolyte disorders, the severity of the clinical manifestations of hypercalcemia depends on the acuteness of onset. Hypercalcemia can be a medical emergency, especially when serum concentrations rise above 14 mg/dL (>3.5 mmol/L). Symptoms associated with this condition often consist of vague GI complaints such as nausea, vomiting, abdominal pain, dyspepsia, and anorexia. More severe GI complications include peptic ulcer disease, possibly due to increased gastrin release, and acute pancreatitis.43,44
Severe hypercalcemic symptoms primarily involve the neuromuscular system (e.g., lethargy, obtundation, psychosis, cerebellar ataxia, and, in severe cases, coma and death). However, EKG changes and spontaneous ventricular arrhythmias may also be seen. It may also enhance the inotropic effects of digoxin, increasing the likelihood of cardiac arrhythmias.35-38
Renal function may be affected by hypercalcemia through the ability of calcium to inhibit the adenyl cyclase–cyclic adenosine monophosphate system that mediates the ADH effects on the collecting ducts. This inhibition results in diminished conservation of water by the kidneys. The renal effect is further compounded by diminished solute transport in the loop of Henle, leading to polyuria, nocturia, and polydipsia.28 Other chronic renal manifestations include nephrolithiasis, nephrocalcinosis, chronic interstitial nephritis, and renal tubular acidosis.
In addition, hypercalcemia can cause vasoconstriction of the renal vasculature, resulting in a decrease in renal blood flow and GFR. If hypercalcemia is allowed to progress, oliguric acute renal failure may ensue.28 In the presence of high calcium–phosphate product, soft-tissue calcification by the calcium–phosphate complex may occur.
The signs and symptoms described above are mostly seen in patients with severe hypercalcemia. With serum concentrations less than 13 mg/dL (3.2 mmol/L), most patients should be asymptomatic.
Causes of spurious laboratory results. False hypercalcemia can occur if the tourniquet is left in place too long when the blood specimen is drawn. This results from increased plasma-protein pooling in the phlebotomized arm. Falsely elevated calcium should be suspected if serum albumin is greater than 5 g/dL (>50 g/L). Table 6-4 contains the normal range values for tests related to calcium metabolism.
Phosphate
Normal range: 2.3–4.7 mg/dL or 0.74–1.52 mmol/L for adults
Many of the factors that influence serum calcium concentrations also affect serum phosphate, either directly or indirectly. Laboratory values for calcium and phosphate should, therefore, be interpreted together. Since phosphate exists as several organic and inorganic moieties in the body, some clinical laboratories simply report the phosphate value as phosphorus.
Physiology
Phosphate is a major intracellular anion with several functions. It is important for intracellular metabolism of proteins, lipids, and carbohydrates, and it is a major component in phospholipid membranes, RNAs, nicotinamide diphosphate (an enzyme cofactor), cyclic adenine and guanine nucleotides (second messengers), and phosphoproteins. Another important function of phosphate is in the formation of high-energy bonds for the production of ATP, which is a source of energy for many cellular reactions. Phosphate is a component of 2,3-diphosphoglycerate (2,3-DPG), which regulates the release of oxygen from hemoglobin (Hgb) to tissues. In addition, phosphate has a regulatory role in the glycolysis and hydroxylation of cholecalciferol. It is also an important acid–base buffer.35,40
A balanced diet for adults usually contains about 800–1500 mg/day of phosphate. About two-thirds is actively absorbed from the small intestine. Some of the phosphate is absorbed passively with calcium and some is absorbed under the influence of 1,25-DHCC, which also increases the intestinal absorption of calcium. However, phosphate is the first of the two to be absorbed.40
Phosphate absorption is diminished when a large amount of calcium or aluminum is present in the intestine due to the formation of insoluble phosphate compounds. Such large amounts of calcium and aluminum may result from the consumption of antacids. In fact, for patients with CKD who have high serum phosphate concentrations, calcium- and aluminum-containing antacids may be given with meals as phosphate binders to reduce intestinal phosphate absorption.45 It should be noted that due to concerns of detrimental accumulation of aluminum in the CNS as well as the ability to worsen anemia and bone disease, chronic use of aluminum-containing antacids should be avoided.
Phosphate is widely distributed in the body throughout the plasma, extracellular fluid, cell membrane structures, intracellular fluid, collagen, and bone. Bone contains 85% of the phosphate in the body. About 90% of plasma phosphate is filtered at the glomeruli, and the majority is actively reabsorbed at the proximal tubule. Some reabsorption also takes place in the loop of Henle, distal tubules, and possibly the collecting ducts.31 The amount of renal phosphate excretion is, therefore, the amount filtered minus the amount reabsorbed. Increased urinary phosphate excretion can result from an increase in plasma volume and the action of PTH, which can block phosphate reabsorption throughout the nephron. In contrast, vitamin D3 and its metabolites can directly stimulate proximal tubular phosphate reabsorption. In all, 90% of eliminated phosphate is excreted renally while the remainder is secreted into the intestine.31,35,46 Renal handling of phosphate, especially the proximal tubules, therefore, plays an important role in maintaining the homeostatic balance of phosphate. Renal phosphate transport is active, saturable, and dependent on pH and sodium ion. However, fluctuation in serum phosphate mostly results from changes in either the GFR or the rate of tubular reabsorption.2,31,35
Serum phosphate and calcium concentrations as well as PTH and vitamin D levels are intimately related with each other. Serum phosphate indirectly controls PTH secretion via a negative feedback mechanism. With a decrease in the serum phosphate concentration, the conversion of vitamin D3 to 1,25-DHCC increases (which increases serum concentrations of both phosphate and calcium). Both the intestinal absorption and renal reabsorption of phosphate are increased. The concomitant increase in serum calcium then directly decreases PTH secretion. This decrease in serum PTH concentration permits a further increase in renal phosphate reabsorption.30,31
A true phosphate imbalance may result from an abnormality in any of the previously discussed mechanisms and hormones for maintaining calcium and phosphate homeostasis. They may include altered intestinal absorption, altered number or activity of osteoclast and osteoblast cells in bone, changes in renal calcium and phosphate reabsorption, and IV infusions of calcium or phosphate salts.35,40
Hypophosphatemia indicates a serum phosphate concentration of less than 2.3 mg/dL (<0.74 mmol/L). The following three clinical conditions are common causes of decreased serum phosphate concentrations:
- Increased renal excretion40,47,48
- Intracellular shifting
- Decreased phosphate or vitamin D intake41,47,48
To identify the etiology of hypophosphatemia, the serum and urine phosphate concentrations should be evaluated simultaneously. Low urine and serum phosphates indicate either a diminished phosphate intake or excessive use of phosphate-binders. An increased urine phosphate suggests either hyperparathyroidism or renal tubular dysfunction. If the increased urine phosphate is accompanied by elevated serum calcium, the presence of primary hyperparathyroidism or decreased vitamin D metabolism must be considered.
Common causes. Hypophosphatemia commonly results from decreased renal reabsorption or increased GFR, shift of phosphate from extracellular to intracellular fluid, alcoholism, or malnutrition. Phosphate is added to total parenteral nutrition solutions for muscle growth and replenishment of hepatic glycogen storage in malnourished patients. The infusion of concentrated glucose solution increases insulin secretion from the pancreas, which facilitates glucose and phosphate cell entry. Phosphate is used to form phosphorylated hexose intermediates during cellular utilization of glucose. An inadequate phosphate content in these nutritional fluids can decrease anabolism, glycolysis, and ATP and 2,3-DPG production.48
Infusion of concentrated glucose solutions, especially when accompanied by insulin, can produce hypophosphatemia through intracellular phosphate shifting. This condition, known as refeeding syndrome, can occur when an inadequate amount of phosphate is given during total parenteral nutrition (i.e., when a large amount of phosphate is taken up by the newly produced cells during anabolism).
Hypophosphatemia can also occur during treatment of hyperkalemia with insulin and dextrose. In addition, aluminum- and calcium-containing antacids, as well as magnesium hydroxide, are potent binders of intestinal phosphate.45 Overuse of these agents can severely reduce serum phosphate concentrations in patients with normal renal function. Moreover, calcitonin, glucagon, and beta-adrenergic stimulants can decrease serum phosphate concentrations. Thiazide and loop diuretics can increase renal phosphate excretion. However, this effect is often insignificant clinically in otherwise healthy individuals.
Other conditions known to cause hypophosphatemia include nutritional recovery after starvation, treatment of diabetic ketoacidosis, decreased absorption or increased intestinal loss, alcohol withdrawal, the diuretic phase of acute tubular necrosis, and prolonged respiratory alkalosis. To compensate for respiratory alkalosis, carbon dioxide shifts from intracellular to extracellular fluid. This shift increases the intracellular fluid pH, which activates glycolysis and intracellular phosphate trapping. Metabolic acidosis, in contrast, produces a minimal change in serum phosphate.
Uncommon causes. Burn patients often retain a great amount of sodium and water. During wound healing, diuresis often ensues, which results in a substantial loss of phosphate. Since anabolism also occurs during recovery, hypophosphatemia may be inevitable without proper replacement. A moderate reduction in serum phosphate can occur from prolonged nasogastric suctioning, gastrectomy, small bowel or pancreatic disease resulting in malabsorption, and impaired renal phosphate reabsorption in patients with multiple myeloma, Fanconi syndrome, heavy-metal poisoning, amyloidosis, and nephrotic syndrome.47,48
Severe hypophosphatemia. Severe phosphate depletion (<1 mg/dL or <0.32 mmol/L) can occur during diabetic ketoacidosis. The resultant acidosis mobilizes bone, promotes intracellular organic substrate metabolism, and releases phosphate into the extracellular fluid. The glycosuria and ketonuria caused by diabetic ketoacidosis results in an osmotic diuresis that increases urinary phosphate excretion. The combined effects of these events may produce a normal serum phosphate concentration with severe intracellular deficiency. When diabetic ketoacidosis is corrected with insulin, phosphate accompanies glucose to move intracellularly. Serum phosphate is usually reduced within 24 hours of treatment. As the acidosis is corrected, there is further intracellular shifting of phosphate to result in profound hypophosphatemia. The accompanying volume repletion may exacerbate the hypophosphatemia further.
Clinical diagnosis. Patients with a moderate reduction in serum phosphate (2–2.3 mg/dL or 0.64–0.74 mmol/L) are often asymptomatic. Neurological irritability may occur as the serum phosphate concentration drops below 2 mg/dL (<0.64 mmol/L). Severe hypophosphatemia is often associated with muscle weakness, rhabdomyolysis, paresthesia, hemolysis, platelet dysfunction, and cardiac and respiratory failure.
Central nervous system effects often include encephalopathy, confusion, obtundation, seizures, and ultimately, coma. The mechanism for these effects may involve decreased glucose utilization by the brain, decreased brain cell ATP, or cerebral hypoxia from increased oxygen-Hgb affinity, secondary to diminished erythrocyte 2,3-DPG content. This decreased content results in decreased glycolysis, which leads to decreased 2,3-DPG and ATP production. The decreased contents of 2,3-DPG and ATP result in an increased affinity of Hgb for oxygen, eventually leading to decreased tissue oxygenation. The ensuing cerebral hypoxia may explain the persistent coma often seen in patients with diabetic ketoacidosis. Hemolysis may occur, but it is rarely seen at serum phosphate concentrations greater than 0.5 mg/dL (>0.16 mmol/L).
Hyperphosphatemia
Hyperphosphatemia indicates a serum phosphate concentration greater than 4.7 mg/dL (>1.52 mmol/L). There are three basic causes for elevated serum phosphate concentrations:
- Decreased renal phosphate excretion
- Shift of phosphate from intracellular to extracellular fluid
- Increased intake of vitamin D or phosphate-containing products (orally, rectally, or intravenously)
Elevated phosphate concentrations may also result from reduced PTH secretion, increased body catabolism, and certain malignant conditions (e.g., leukemias and lymphomas).4,47,48
Causes. The most common cause of hyperphosphatemia is renal dysfunction, which commonly occurs as the GFR falls below 25 mL/min. Chronic kidney disease results in secondary hyperparathyroidism, which can further reduce renal phosphate elimination. The increase in serum phosphate concentration increases the risk for deposition of insoluble calcium–phosphate complex in soft tissues (i.e., metastatic calcification). This deposition may further reduce the serum concentration of ionized calcium and lead to increased PTH production and release. A sustained period of high PTH level leads to excessive bone resorption, which will severely weaken its structural integrity.36,40
Hyperphosphatemia can be caused by a shift of phosphate from intracellular to extracellular fluid. This shift of phosphate can result from massive cell break down after administering chemotherapy for leukemia or lymphoma, and during rhabdomyolysis and septic shock. In addition, hyperthyroidism can elevate serum phosphate by directly increasing renal tubular phosphate reabsorption.
Clinical diagnosis. Signs and symptoms of hyperphosphatemia commonly result from the accompanying hypocalcemia and hyperparathyroidism (see Hypocalcemia section). Renal function may diminish if hyperphosphatemia is left untreated. In the presence of renal dysfunction, phosphate excretion is further reduced to cause an even greater increase of serum phosphate concentration and a further decline in serum calcium concentration (Minicase 4).36,40,45
Calcium and Phosphate Disorders in a Patient with Chronic Renal Failure
MICHAEL S., A 65-YEAR-OLD MAN, had a 1-week history of nausea, vomiting, and general malaise. His appetite had severely decreased over the past 2 months. He had a longstanding history of uncontrolled hypertension and type 2 diabetes mellitus as well as diabetic nephropathy, retinopathy, and neuropathy.
His current medications include levothyroxine 0.1 mg orally daily, metoclopramide 10 mg orally 3 times a day, and a subcutaneous insulin regimen given twice a day. His physical examination revealed a BP of 160/99 mm Hg, diabetic retinopathic changes with laser scars bilaterally, and diminished sensation bilaterally below the knees.
His laboratory values were serum sodium 146 mEq/L (136–142 mEq/L), potassium 4.7 mEq/L (3.8–5.0 mEq/L), chloride 104 mEq/L (95–103 mEq/L), total carbon dioxide content 15 mmol/L (24–30 mmol/L), SCr 3.2 mg/dL (0.6–1.2 mg/dL), BUN 92 mg/dL (8–23 mg/dL), and random blood glucose of 181 mg/dL (70–110 mg/dL). Because of his renal failure, additional laboratory tests were obtained: calcium 7.5 mg/dL (9.2–11.0 mg/dL), phosphate 9.1 mg/dL (2.3–4.7 mg/dL), albumin 3.3 g/dL (3.5–5 g/dL), and uric acid 8.9 mg/dL (4.0–8.5 mg/dL).
Over the next several days, he complained of finger numbness, tingling, and burning of extremities. He also experienced increasing confusion and fatigue. A neurological examination was positive for both Chvostek and Trousseau signs. Repeated laboratory tests showed substantial changes in serum calcium (6.1 mg/dL) and phosphate (10.4 mg/dL). The patient’s intact serum PTH was 280 pg/mL (10–65 pg/mL).
Question: What calcium and phosphate disorders does Michael S. have?
Discussion: Michael S. has three laboratory abnormalities that are related specifically to calcium–phosphate metabolism: (1) hypocalcemia, (2) hyperphosphatemia, and (3) hyperparathyroidism. He is exhibiting classic signs and symptoms of hypocalcemia, such as finger numbness, tingling, burning of extremities, confusion, fatigue, and positive Chvostek and Trousseau signs.
Chronic kidney disease, as seen in Michael S., is commonly associated with hypocalcemia, hyperphosphatemia, hyperparathyroidism, and vitamin D deficiency. These calcium–phosphate abnormalities are responsible for the development of renal osteodystrophy. During the early stages of renal failure, renal phosphate excretion began to decrease. His serum phosphate concentration was thus increased and the ionized calcium concentration became reduced, which stimulated the release of PTH, resulting in secondary hyperparathyroidism. The higher concentration of PTH reduced his renal tubular phosphate reabsorption, thereby increasing its excretion. The hyperparathyroidism thus helped to maintain his serum phosphate and calcium concentrations within normal ranges during the early stage of renal failure (Figure 6-3).
As renal function continues to deteriorate (GFR below 30 mL/min), renal tubules ceased to respond adequately to the high serum PTH concentration, resulting in hyperphosphatemia. In response to the hypocalcemia that followed, calcium was mobilized from the bone through the action of PTH. However, such compensatory response is not sufficient as hypocalcemia and hyperphosphatemia continued. The persistent hyperphosphatemia may contribute to the diminished renal conversion of 25-HCC to its biologically active metabolite 1,25-DHCC. As a result, the gut absorption of dietary calcium was diminished. Deficiency in active vitamin D can therefore aggravate his hypocalcemia, which subsequently can stimulate PTH secretion increasing mobilization of calcium from bone. The metabolic acidosis that is common in renal failure may also contribute to the negative calcium balance in the bone.
Michael S. was relatively asymptomatic up to this point, primarily because these laboratory abnormalities were developed over a long period of time and therefore allowed the body to compensate. In the presence of nausea and vomiting and the lack of appetite, his oral calcium intake was probably reduced substantially, which might have enhanced his malaise. Since calcium is commonly reported as total calcium and not as the free or ionized fraction, his total serum calcium concentration must be corrected for his low serum albumin value. For every 1 g/dL reduction in serum albumin below 4 g/dL, 0.8 mg/dL should be added to his serum calcium concentration. Therefore, with his serum albumin concentration of 3.3 g/dL, his initial serum calcium value of 7.5 mg/dL is equivalent to a total calcium concentration of about 8.1 mg/dL. Therefore, he does have true hypocalcemia, although the deficit is mild.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


