57 Dystrophia myotonica
Salient features
History
• Onset usually in the third and fourth decade. However, if the mother is the carrier then the disease may manifest in infancy and undergo rapid deterioration at the usual age of onset
• Onset dominated by weakness or myotonia or both
• Difficulty in releasing grip
• Leg weakness (difficulty in kicking a ball)
• ‘Pseudo-drop attacks’ (caused by weakness of quadriceps muscle)
• Ask the patient if they have dysphagia (oesophageal involvement)
• Impotence (caused by gonadal atrophy)
• Recurrent respiratory infections (caused by weakness of muscles of bronchioles)
Examination
• While shaking hands with the patient, note the myotonia
• Frontal baldness (the patient may be wearing a wig and it is important to mention if this is so)
• Ptosis (bilateral or unilateral) with a smooth forehead
• Cataracts (posterior capsular cataracts) or evidence of surgery for cataracts
• Difficulty in opening the eyes after firm closure
• Expressionless face (‘hatchet face’; Fig. 57.1) with wasting of temporalis, masseters and sternomastoids and ‘swan neck’ caused by thinning of the neck.
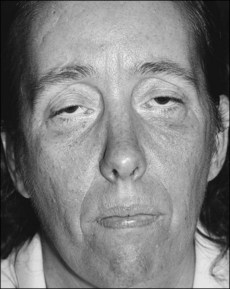
Fig. 57.1 Muscle wasting in myotonic dystrophy gives the characteristic drawn appearance of ‘hatchet facies.’
(With permission from Yanoff, Duker 2008.)
Remember: Respiratory failure is the commonest cause of death.



