Dyslipidemia
LEARNING OBJECTIVES
• INTRODUCTION
In 2011, Centers for Disease Control estimated that 29% of all deaths in the U.S. will be caused by heart and cerebrovascular disease, the vast majority of which are caused by atherosclerotic cardiovascular disease (ASCVD), also known as coronary heart disease (CHD). Atherosclerosis is a process that causes oxidative endothelial damage and inflammation that eventually leads to the formation of cholesterol-laden atheromas or plaque lining the arterial intima. The inflammation ultimately causes the plaque to rupture, which leads to platelet adhesion and aggregation resulting in arterial occlusion. If vessels in the heart and brain are occluded they cause a myocardial infarction or cerebrovascular accidents (strokes or CVA). Since dyslipidemia is a major factor in the atheroscerlotic process, the advent of highly effective cholesterol-lowering medication has become a major tool in preventing atherosclerosis and subsequent heart attacks and strokes. Pharmacists play a key role in monitoring and managing the therapeutic regimens of patients with dyslipidemia.
• TYPES OF LIPIDS
There are three major types of cholesterol found in the bloodstream. They are carried in the blood by lipoproteins, which combine with cholesterol and other fats. LDL cholesterol (low-density lipoprotein or LDL-C) is the cholesterol intimately involved in the formation of arterial plaque and high levels are associated with a greater risk of heart attack and stroke. Since lowering LDL-C blood concentration has been shown to prevent heart attacks and strokes, it is the focus of current lipid-lowering therapy. LDL-C is made by the liver from VLDL cholesterol (very-low-density lipoprotein or VLDL-C), which has the highest concentration of triglycerides. HDL cholesterol (high-density lipoprotein or HDL-C) serves as a scavenger, transporting LDL-C back to the liver for recycling, and maintaining the integrity of the endothelium of the arteries. High levels of HDL-C markedly reduce the incidence of heart attacks and strokes, so increased HDL-C blood levels reduce the risk of cardiovascular disease. Some authors refer to LDL as the “bad” cholesterol and the HDL as the “good” cholesterol. These terms can be very useful in helping patients understand their illness and the importance of treatment goals, i.e., lowering the bad cholesterol and raising the good. Triglycerides are a fourth type of lipid routinely found in the blood stream. They are important sources for energy, but high levels are associated with the metabolic syndrome and its increased risk for cardiovascular disease and type 2 diabetes mellitus.
• DIAGNOSING DYSLIPIDEMIA AND THE NEED FOR THERAPY
According to the 2002 guidelines (National Cholesterol Education Program Adult Treatment Panel III or NCEP ATP Panel III), diagnosis and the determination of need for therapy were both tied to a risk factor assessment, based in part on LDL-C levels. Normal LDL-C values vary based on risk factors and HDL-C levels. To determine what a normal LDL-C is for an individual, which was also their target LDL-C goal, a series of questions needed to be asked and calculations done (Table 19.1). There are significant controversies surrounding the new, 2013 guidelines and numerous experts have suggested continuing to use the old guidelines for the time being. Therefore, the old criteria and cases to practice calculating older cardiovascular risk and target goals of therapy based on LDL-C are included to enable pharmacists to work with physicians who continue to utilize older guidelines.
| TABLE 19.1 | Low-Density Lipoprotein (LDL) Goal Calculation (NCEP ATP III) |

The new, 2013 guidelines eliminate the use of LDL-C target goals! Instead the new guidelines focus on cardiovascular risk, advocating reduction of LDL-C levels by 30% to 50% by choosing the dose and potency of the statin (Table 19.2). The Framingham-based ASCVD risk calculator from NCEP ATP III has been replaced by the new Pooled Cohort Equations Cardiovascular Risk Calculator found at http://myamericanheart.org/cvriskcalculator. Table 19.2 shows the four categories of patients that will benefit from statin therapy. They include those with clinical ASCVD and those with LDL-C levels higher than 190 mg/dL. In addition, patients with diabetes, without clinical ASCVD, but with LDL-C levels of 70 to 189 mg/dL, plus those without diabetes and clinical ASCVD, LDL-C levels 70 to 189 mg/dL, but with a 10-year ASCVD risk of >7.5%, also warrant statin therapy. See Table 19.2 or Key reference 15 for details about the appropriate intensity of statin therapy for each group. Dosage titration of statins to target LDL-C goals has been eliminated. Nonstatin medication use is discouraged.
| TABLE 19.2 | 2013 ACC/AHA Cholesterol Guidelines |

• OTHER RISK FACTOR MEASUREMENTS
While the focus of the new guidelines is on LDL-C levels, there are other biomarkers that can be used to further assess the risk of developing ASCVD (Table 19.3).
| TABLE 19.3 | Supplementary Factors Indicating Increased ASCVD Risk |
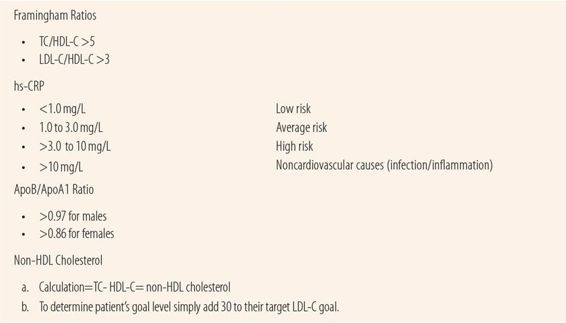
Framingham Study Ratios
The Framingham Study began in 1948 and has traced more than 15,000 patients over their lifetimes to see what risk factors exist for developing cardiovascular diseases of all types. Today, much of the data that we use to set practice standards for most cardiovascular diseases came from the Framingham Study. The study results established ratios of cholesterol values that were definitely linked to markedly increased risk of ASCVD. They found major risks associated with two ratios using HDL-C. If total cholesterol value (TC) divided by the HDL-C (TC/HDL) was >5 or the LDL-C divided by the HDL-C (LDL/HDL) was >3, there was a markedly increased risk of coronary artery disease. These can be useful today in patients with borderline risk profiles to determine whether or not there is significant risk of ASCVD that warrants initiation of cholesterol-lowering therapy. For example, a male patient who is 40, but has hypertension and an HDL-C of 22, a total cholesterol of 239, LDL-C = 150 and smokes, has three risk factors: smoking, low HDL-C, and hypertension. However, he is planning to quit smoking with a quit date next week. He has already begun taking bupropion. If the patient stays quit for 30 days, that would eliminate smoking as a risk factor, giving him just two risk factors under the 2002 guidelines. Assuming his systolic pressure runs between 135 and 145 mm, the Framingham Score Calculation Tool would give him a 10-year risk of heart attack of 12% and an LDL-C target of <130 mg/dL. However, his TC/HDL ratio is well above 5 (10.9) and his LDL/HDL ratio is well above 3 (6.8), indicating major risks for cardiovascular disease. In this case, in addition to quitting smoking, he should be counseled about his risk and advised to increase his exercise and modify his diet to raise his HDL-C above 40 mg/dL and to lower his total and LDL-C cholesterol. The new, 2013 guidelines make no reference to Framingham ratios.
Apolipoprotein B/Apolipoprotein A1 Ratio
Apolipoprotein B is the transporter for LDL-C and binds to the receptors on the cells facilitating deposit. Apolipoprotein A1 plays a similar role for HDL-C. Recent evidence indicates that this ratio is a more accurate marker of risk than the LDL/HDL ratio. An ApoB/ApoA1 ratio of >0.97 for men and >0.86 for women is indicative of increased risk of ASCVD. Additional evidence appears to make it more accurate in subclinical atherosclerosis and may be a better predictor of plaque instability than traditional lipid profiles. Recent studies have shown that a formula to estimate ApoB/ApoA1 ratios, using only HDL and total cholesterol, has excellent correlation with direct measurement in fasting patients. The new, 2013 guidelines make no reference to this ratio.
Non-HDL Cholesterol
Some experts feel that non-HDL cholesterol should be the primary parameter used rather than LDL-C alone since it better predicts the risk of developing ASCVD. One reason is because the LDL-C value is an estimate based on the Friedewald formula using HDL, total cholesterol, and triglycerides and does not include VLDL-C, which is also atherogenic. High triglyceride levels make the estimate much less reliable, hence when testing patients with high triglyceride levels the report may say “unable to calculate” for LDL-C values. The 2002 guidelines accommodated those experts by suggesting the use of non-HDL cholesterol as a secondary goal after current LDL-C goals are met. Target non-HDL-C goals can be calculated by adding 30 mg/dL to each of the three LDL-C levels derived from risk-factor assessment.
C-Reactive Protein High-Sensitivity Assay
Because endothelial inflammation and subsequent plaque rupture are such a major part of the atherosclerotic process, many experts have advocated the use of C-reactive protein high-sensitivity assay (hs-CRP) as an adjunctive marker of ASCVD risk. C-reactive protein is a biomarker of inflammatory activity in the body. Its release from the liver is stimulated by several cytokines including IL-1 and IL-6. CRP stimulates the production of cytokines and chemokines in endothelial cells contributing to the inflammatory endothelial dysfunction that is part of the atherosclerotic process. The American Heart Association and CDC recommend that in asymptomatic patients with a 10% to 20% 10-year risk of myocardial infarction hs-CRP be used to further assess CHD risk using the following criteria. An hs-CRP below 1.0 mg/L predicts low risk; an hs-CRP level between 1.0 and 3.0 mg/L indicates average risk, whereas hs-CRP levels above 3.0 mg/L indicate a high risk of CHD. Persistent levels >10 mg/L indicate noncardiovascular causes like an acute infection or inflammation. The JUPITER study showed that rosuvastatin reduced cardiovascular risk by 37% in patients with low/normal cholesterol levels but high hs-CRP levels. Recent studies have shown a genetic variation in hs-CRP levels further adding to the controversy surrounding the accuracy and utility of hs-CRP as a biomarker of cardiovascular disease. The new guidelines use an hs-CRP of 2 mg/L or greater as a potential factor to justify starting stains in patients at very low risk of ASCVD.
• MONITORING/MANAGING DYSLIPIDEMIA
Therapeutic lifestyle changes (low fat diets and exercise) are the mainstay in the treatment of dyslipidemia. However, the majority of patients do not achieve the degree of LDL-C reduction they need from just lifestyle changes. Statins have been shown to markedly reduce the risk from ASCVD. There are two major mechanisms of action that account for this benefit. First, they reduce the synthesis of LDL-C, a major component of atheromatous plaque. Second, they have an anti-inflammatory effect that decreases endothelial reactivity and ultimately reduce the tendency for plaque rupture. Recent studies have shown that adding other cholesterol-lowering agents to statins improves lipid profiles, but has no effect on cardiovascular outcomes. Even though hs-CRP levels are lowered by statins, there is little or no evidence regarding dose-effect relationship for the anti-inflammatory effect. Therefore, cardiovascular risk factors are still used to determine whether or not to start statin therapy.
Monitoring Treatment Efficacy
The lipid profile (total cholesterol, HDL-C, LDL-C, and triglycerides) is the primary measure of therapy effectiveness. From those values, the Framingham ratios (TC/HDL, LDL/HDL) and non-HDL cholesterol levels (TC level minus HDL-C value) can be calculated. ApoB/ApoA1 ratios and hs-CRP must be ordered separately at this time. In patients who choose to try therapeutic lifestyle changes alone as initial therapy, a trial of 6 months is usually appropriate. Lipid profiles are done at baseline and 4 to 12 weeks after initiation of statin therapy and at least annually and up to every 3 months. This 4- to 8-week period allows the statin dose to attain its maximum impact. In the patient attempting therapeutic lifestyle changes alone, repeating the lipid profile every 4 to 8 weeks, over the 6 months, can serve as educational and motivational tools, as well as provide data to the reluctant patient to help them see the need for statin therapy, if appropriate, in the future.
Under the 2002 guidelines, “When do I increase statin doses or add nonstatin medication?”, was a frequent concern of providers. Guidelines by definition are not absolute requirements, but suggestions based on the best available evidence; therefore, application of common sense is important. As with hypertension, be careful not to “chase numbers.” An LDL-C level of 5 mg/dL above the patient’s target goal does not mandate an increase in medication dose. As in a patient with hypertension, in an asymptomatic patient near LDL-C target goal, it is perfectly appropriate to wait another 6 months to get another lipid profile. Waiting can serve as motivation for the patient to further improve the intensity of their lifestyle changes. In patients with dyslipidemia, the alternate measurements of risk can assist in the decision regarding potential dose increases. For example, a patient close to target LDL-C goal, but with unfavorable Framingham or ApoB/ApoA1 ratios, and an hs-CRP level >3.0 mg/L would be a more viable candidate for a dose increase. Conversely, in a patient with the same LDL-C levels and favorable alternative parameters, the pharmacist could more comfortably delay a dosage increase. It is not uncommon in a patient with dyslipidemia and type 2 diabetes to have high triglycerides in spite of LDL cholesterol levels below 100 mg/dL. Since excess glucose is the major source of triglycerides, the patient’s elevated triglycerides are most likely due to poor control of their diabetes and increasing the statin dose or adding an agent to lower triglycerides, e.g., fibrates would be inappropriate until the patient’s A1C is below 7.0. Therefore, in this situation, intensifying therapy for their type 2 diabetes is the treatment of choice to lower the triglyceride level, especially considering that concurrent fibrate therapy markedly increases the risk of musculoskeletal adverse effects of statins, specifically rhabdomyolysis.
Under the new guidelines, if a patient does not meet expected percentage providers are told to check for adherence to statins and lifestyle modifications. There is no provision for increasing statin doses unless the patient moves into a different risk-benefit group, by developing diabetes, having a cardiovascular event, or having their LDL-C levels exceed 190 mg/dL. While all patients taking statins should be monitored for adherence, in patients taking statins other than pitavastatin, atorvastatin, or rosuvastatin, who are apparently not responding to statin therapy with sufficient LDL-C lowering, providers need to verify that the patients are taking their statin after 6 pm in the evening before changing therapy. Many purported cases of ineffectiveness of simvastatin and pravastatin are, in part, due to inappropriate timing of the dose.
• MONITORING FOR COMPLICATIONS OF DYSLIPIDEMIA AND ITS TREATMENT
Disease Complications
Complications of dyslipidemia are those of atherosclerosis: angina pectoris, acute coronary syndrome, peripheral artery disease (intermittent claudication), and stroke (CVA). Therefore, questions about the symptoms of those complications are appropriate at each visit, e.g., chest pain, leg pain when exercising, visual disturbances (Table 19.4). Since dyslipidemia frequently occurs concurrently with hypertension and diabetes, which also cause the same atherosclerotic complications, it is unnecessary to ask questions for each disease, i.e., asking about chest pain once covers common complications for all three comorbid conditions.
| TABLE 19.4 | Follow-Up Visit Check List for Dyslipidemia |

Drug Complications
Adverse drug reactions with statins are uncommon. Musculoskeletal disorders and hepatic side effects have been prominent concerns historically. Recently, concerns have been raised regarding statin-induced cognitive adverse effects and a statin-related increased incidence of diabetes.
Musculoskeletal Myalgias occurred in 1.5% to 3.0% of patients in randomized controlled trials of statins. Observational studies have shown a slightly higher incidence, up to 10%. Patients describe symptoms including pain, tenderness, cramping, fatigue, weakness, stiffness, and heaviness. Symptoms are symmetrical (occurring in limbs on both sides of the body). Muscle symptoms tend to be generalized and proximal (occurring in both upper and lower limbs) and may be worse with exercise. In general, the higher the dose of statin, the more likely muscle symptoms are to occur. Reactions most commonly occur within a month of initiation or dosage increase, but in one study, 15% reported symptom onset after 6 months of therapy. Generally, symptoms resolve within 2 to 8 weeks after discontinuation.
Statins have rarely caused rhabdomyolysis, an acute fulminant, potentially fatal disease of skeletal muscle, involving the destruction of muscle manifested by myoglobinemia, myoglobinuria, brown urine, and eventually renal failure. The annual incidence of statin-induced rhabdomyolysis is 0.0042% (4.2/100,000) with statin monotherapy. Concurrent therapy with fibrates increases the risk of rhabdomyolysis to 200/100,000. Symptoms include diffuse, symmetrical muscle pain and weakness, flu-like symptoms, and low back pain. Creatine kinase (CK) levels are at least 10 times the upper limit of normal, and myoglobin is present in the urine. Published criteria include elevated serum creatinine, but some experts feel that is not necessary for the diagnosis.
The cause of statin muscle side effects is unknown, but has been attributed to effects on coenzyme Q10, selenoproteins, and low cholesterol itself. Multiple studies have failed to support any individual cause. Patient education should include warnings about symmetrical, diffuse pain, or weakness in the extremities and at each visit patients should be questioned about those symptoms. If a patient complains of musculoskeletal pain/weakness typical of statins, then a careful history should be taken, which includes looking for other medications that may interfere with the metabolism of statins by the liver or other conditions that might increase statin levels (liver or renal disease, depending on the statin). CK serum levels should be drawn. Thyroid-stimulating-hormone (TSH) levels should also be drawn since hypothyroid disease raises serum cholesterol and CK levels, and may predispose patients to statin myotoxicity. Some authors also suggest 25-OH vitamin D levels because vitamin D deficiency has been associated with statin myotoxicity. Obviously, the presence of clinical and laboratory evidence of rhabdomyolysis requires immediate discontinuation of the statin and treatment. Normal CK levels rule out muscle damage and the decision to discontinue the statin depends on the tolerability of muscle symptoms compared to the level of need for cholesterol reduction. More frequent CK serum levels are indicated to quickly identify any progression of the myotoxicity. Muscle symptoms plus elevated CK levels of less than 10 times the upper limit of normal are more problematic. Guidelines suggest that cautious continuation with frequent clinical and laboratory monitoring can be appropriate. Lower doses or switching to a less lipophilic statin (pravastatin or rosuvastatin) have been suggested, but there are no clinical trials to support their efficacy. Intolerable muscle symptoms, like rhabdomyolysis, require immediate discontinuation. If discontinuation does not resolve muscle symptoms, then a more detailed workup, potentially including muscle biopsy, is indicated.
Hepatotoxicity Hepatotoxicity due to statins has been a controversial issue. Based on reports of increased transaminase levels in early trials, the FDA originally required baseline liver function tests (LFTs), followed by repeat LFTs every 6 months. However, recent evidence has led to uncertainty regarding the need for monitoring with LFTs. First, while there have been cases of acute liver failure thought to be caused by statins they have been confounded by the presence of other hepatotoxic agents. In addition, the rate of purported statin-induced acute hepatic failure is less than that of liver failure due to idiosyncratic causes. Second, nonalcoholic liver disease (NALD) commonly exists in patients with the metabolic syndrome and causes low levels of transaminase elevations. Since dyslipidemia is a major component of the metabolic syndrome, many patients on statins could have elevated LFTs due to the metabolic syndrome, rather than statins, making it problematic to distinguish NALD from the typical statin-induced mild transaminase elevations. Third, several studies have shown that giving statins to patients with mild transaminase elevations, including patients with hepatitis C is safe. In some patients addition of statins actually caused reductions in transaminase levels. Given the additional information, the FDA recently dropped the requirement for both baseline and follow-up LFTs for all statins. However, it is appropriate to question patients for potential signs and symptoms of hepatotoxicity at each visit, i.e., fatigue, malaise, anorexia, nausea, vomiting, jaundice, dark urine. Positive responses should be followed up with LFTs.
Diabetes and Cognitive Dysfunction Recent case reports and epidemiologic studies have proposed potential statin-induced cognitive dysfunction and an increased incidence of diabetes in statin users. Several case reports have suggested cognitive dysfunction as an adverse effect of statins. Symptoms include primarily impaired concentration and attention, or long- and short-term memory loss. However, behavioral changes, anxiety, and paranoia have also been reported. Symptom onset ranges from 5 to 270 days and patient recovery time has ranged from a few days to 4 weeks after discontinuing statins. The FDA MedWatch program received 60 case reports of memory loss attributable to statins over a 5-year period. There have been 11 studies looking at cognitive effects of statins as a primary endpoint. Three studies showed improvement in cognitive function; seven showed no difference between patients receiving statins compared to patients receiving placebo. Only one study showed negative effects on some measures of cognitive function. Furthermore, no significant differences in cognitive function were found between patients on statins compared to those on placebo in three studies where cognitive function was a secondary endpoint. The theoretical basis for statin-induced cognitive dysfunction involves the important role of cholesterol in the brain as part of the myelin sheath, but exact mechanisms are unknown. Since statin-induced cognitive impairment is uncommon to rare, patients should be questioned at each visit for changes in concentration, attention, and memory loss. Also in patients exhibiting behavioral changes, the statin should be included in the differential diagnosis of the etiology of the symptoms. If symptoms are thought to be due to a statin, discontinuing the statin for 1 to 3 months appears to be safe, not increasing the risk of cardiovascular events. In patients who have statin-induced cognitive dysfunction, a discussion of risk versus benefit should take place. Various authors and experts have suggested several therapeutic options for patients with likely statin-induced cognitive issues. Most common is switching the patient to a more hydrophilic statin (pravastatin or rosuvastatin), since almost all reports have occurred with more lipophilic statins (simvastatin, atorvastatin). Some experts, who feel that all their patients with cognitive dysfunction had LDL-C levels markedly below 100 mg/dL, have suggested lowering the dose of statin to allow the LDL-C serum level to rise to near the target goal for patients with higher cardiac risk (100 mg/dL). There are no clinical trials to support the efficacy of either of these theoretical options.
In several meta-analyses, researchers noted an increased incidence of diabetes, especially in those patients over 65 who have a history of long-term statin use. The risk ranged from 9% to 13%. While some studies looked at changes in BMI to rule out the metabolic syndrome as a cause of increased diabetes risk, these studies were, at best, hypothesis generating. Given the genetic predisposition of the metabolic syndrome and type 2 diabetes, and the concurrent presence of dyslipidemia in both conditions, it would not be an unexpected finding for the incidence of diabetes to be higher in patients with abnormal cholesterol values. Until better evidence is available, it is appropriate to monitor fasting plasma glucose or A1C levels periodically in all patients on statins.
• KEY REFERENCES
1. Grundy SM, Becker D, Clark LT, et al. Third Report of the National Cholesterol Education Panel (NCEP) Expert Panel: Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III): Final Report. Circulation. 2002;106:3143-3421.
2. Grundy SM, Cleeman JL, Merz CN, et al. Implications of Recent Clinical Trials for the National Cholesterol Education Programs Adult Treatment Panel III Guidelines. Circulation. 2004;110:229-239.
3. Last AR, Ference JD, Falleroni J. Pharmacological treatment of dyslipidemia. Am Fam Physician. 2011;85:551-558.
4. Rana JS, Boekholdt SM. Should we change our lipid management strategies to focus on non-high-density lipoprotein cholesterol? Curr Opin Cardiol. 2010;26:622-626.
5. Contreras F, Lares M, Castro J, et al. Determination of non-HDL cholesterol in diabetic and hypertensive patients. Amer J Ther. 2010;17:337-340.
6. Robinson JG. LDL reduction: how low should we go and is it safe? Curr Cardiol Rep. 2008;10:481-487.
7. Sniderman AD, De Graaf J, Couture P. Low-density lipoprotein strategies: target versus maximalist versus population percentile. Curr Opin Cardiol. 2012;27:405-411.
8. Battistoni A, Rabattu S, Volpe M. Circulating biomarkers with preventive, diagnostic and prognosic implications in cardiovascular diseases. Int J Cardiol. 2012;157:160-168.
9. Sathasivam S. Statin-induced myotoxicity. Eur J Intern Med. 2012;23:317-324.
10. Mansi I, Frei CR, Pugh MJ, et al. Statins and musculoskeletal conditions, arthropathies and injuries. JAMA Intern Med. 2013;173: 1318-26.
11. Russo MW, Scobey M, Bonkovsky HL. Drug-induced liver injury associated with statins. Semin Liver Dis. 2009;29:412-422.
12. Bader T. The myth of statin-induced hepatotoxicity. Am J Gastroenterol. 2010;105:978-980.
13. Rojas-Fernandez CH, Cameron JCF. Is statin-associated cognitive impairment clinically relevant? A narrative review and clinical recommendations. Ann Pharmacother. 2012;46:549-557.
14. Sampsom UK, MacRae FL, Fazio S. Are statins diabetogenic. Curr Opin Cardiol. 2011;26:342-347.
15. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. [Published online November 12, 2013] Circulation doi:10.1161/01 cir 0000437741.48606.98.
16. Richardson K, Schoen M, French B, et al. Statins and cognitive function: a systematic review. Ann Intern Med. 2013;159:688-697.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


