Chapter 16 Drugs for inflammation and joint disease
Inflammation
Several drugs in current use act on the various stages of this inflammatory process. Antagonists of TNFα, IL-1 and IL-6 are available (see Biologic agents, p. 253). Colchicine, used in the treatment of gout, interferes with neutrophil chemotaxis, thus inhibiting their recruitment to the site of inflammation.
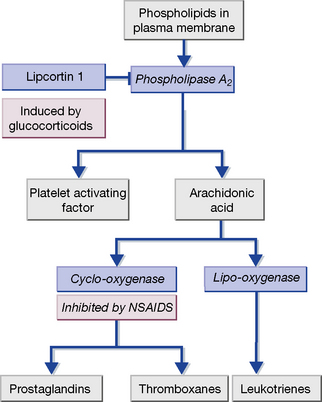
Many leucocytes, including mast cells and macrophages, as well as endothelial cells, synthesise pro-inflammatory eicosanoids and platelet-activating factor (PAF) (Fig. 16.1). These are 20-carbon unsaturated fatty acids derived from phospholipid substrates in the plasma membrane by the enzymes phospholipase A2, cyclo-oxygenase (COX) and lipo-oxygenase (which are induced by IL-1). The prostaglandins, thromboxanes and leukotrienes have diverse pro-inflammatory roles. Leukotrienes promote the activation and accumulation of leucocytes at sites of inflammation. Prostaglandins induce vasodilatation of the microcirculation and are important in pain signalling from locally inflamed tissue. Platelet-activating factor and thromboxane A2 affect the coagulation and fibrinolytic cascades. Non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, inhibit COX and hence prostaglandin and thromboxane synthesis. Glucocorticoids act by inducing the synthesis of lipocortin-1, a polypeptide that inhibits phospholipase A2, and thereby exert a broad anti-inflammatory effect. The leukotriene receptor antagonists montelukast and zafirlukast cause bronchodilatation and are used to treat asthma.
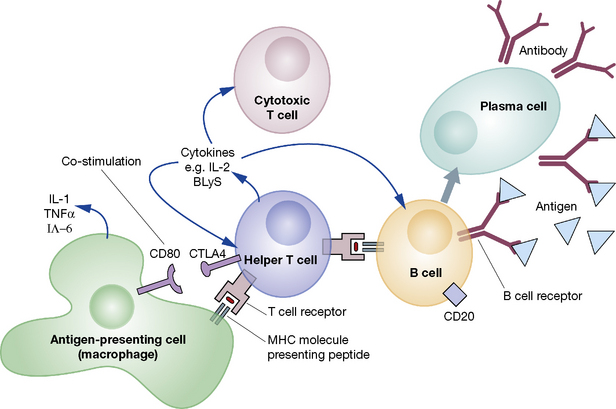
The adaptive immune response
An adaptive immune response is initiated when a helper T cell recognises a peptide antigen presented on the surface of an antigen-presenting cell (APC) and is activated (Fig. 16.2). The activated helper T cell is then able to activate other types of T cell and B cells. This results in the proliferation of adaptive cellular effectors, the generation and release of antibodies by plasma cells and the production of a range of cytokines by the participating leucocytes. On occasion, amplification loops may become self-perpetuating, leading to chronic autoimmune disease.
Pharmacological manipulation of inflammatory mediators
Mode of action
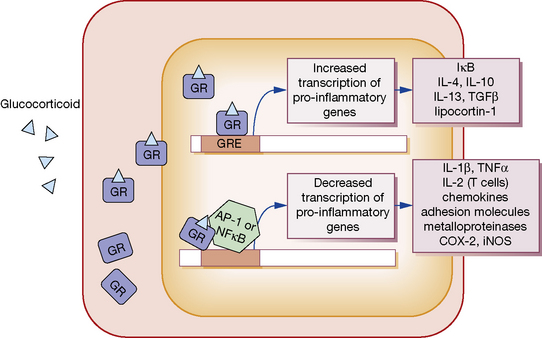
GCs, being lipophilic, diffuse across the cell membrane and bind the cytosolic glucocorticoid receptor (GR) (Fig. 16.3). Receptor polymorphisms influence the strength of the receptor interaction, and represent one source of variation in sensitivity to exogenous steroids. Once bound, the GC-GR complex translocates to the nucleus where it acts in at least two ways to alter gene transcription:
1. The GC-GR complex binds to the glucocorticoid response element within target gene promoters, increasing transcription of various anti-inflammatory genes. These include I-κB, which inhibits the activation of nuclear factor (NF)-κB, and the cytokines IL-4, IL-10, IL-13 and transforming growth factor (TGF)β, which have immunosuppressive and anti-inflammatory activity.
2. The GC-GR complex interferes with the binding of the transcription factors activating protein (AP)-1 and NF-κB to their response elements. This action decreases the transcription of a range of pro-inflammatory mediators. These include IL-1β and TNFα; IL-2, which stimulates T-cell proliferation; a range of chemokines and cellular adhesion molecules; metalloproteinases; COX-2; and inducible nitric oxide synthase.
3. GCs increase synthesis of the polypeptide lipocortin-1, which inhibits phospholipase A2 and thereby the synthesis of prostaglandins, thromboxane A2 and PAF (see Fig. 16.1).
Non-steroidal anti-inflammatory drugs (NSAIDs)
Recently concern has arisen over the effect of traditional NSAIDs and COX-2 inhibitors on the cardiovascular system, with analysis of the VIGOR1 study showing that rofecoxib in particular increases the risk of myocardial infarction (rofecoxib has since been withdrawn from use). While they retain an important role in the treatment of acute gout, inflammatory arthritis, ankylosing spondylitis and dysmenorrhea, long-term prescription should only be undertaken following a full discussion with the patient regarding the balance of risks and benefits.
Mode of action
NSAIDs inhibit cyclo-oxygenase (COX), which catalyses the synthesis of prostaglandins and thromboxane from arachidonic acid (see Fig. 16.1). There are two isoforms of COX:
• COX-1 is constitutively2 expressed in most cell types.
• COX-2 is induced when inflammatory cells (fibroblasts, endothelial cells and macrophages) are activated by cytokines such as IL-1β and TNFα. It is also often upregulated in cancer cells, and is constitutively expressed in the kidney and brain.
NSAIDs can be categorised according to their COX specificity as:
Pharmacokinetics
NSAIDs are absorbed almost completely from the gastrointestinal tract, tend not to undergo first-pass elimination (see p. 87), are highly protein bound and have small volumes of distribution. Their t½ values in plasma tend to group into short (1–5 h) or long (10–60 h). Differences in t½ are not necessarily reflected proportionately in duration of effect, as peak and trough drug concentrations at their intended site of action following steady-state dosing are much less than those in plasma. The vast majority of NSAIDs are weak organic acids and localise preferentially in the synovial tissue of inflamed joints (see pH partition hypothesis, p. 80).
Adverse effects
Gastrointestinal
NSAID-associated gastrointestinal disease appears to result from the inhibition of COX–1-mediated production of cytoprotective mucosal prostaglandins, especially PGI2 and PGE2, which inhibit acid secretion in the stomach, promote mucus production and enhance mucosal perfusion. Several large randomised controlled trials have investigated the incidence of gastrointestinal adverse effects in traditional NSAIDs compared with coxibs. The VIGOR (rofecoxib versus naproxen), CLASS3 (celecoxib versus ibuprofen and diclofenac) and TARGET4 (lumiracoxib versus ibuprofen and naproxen) studies all indicate that coxib use leads to an approximately 50% reduction of upper gastrointestinal adverse events.
Cardiovascular
The VIGOR and APPROVE5 trials reported increased thrombotic cardiovascular events in patients treated with rofecoxib, leading to concerns about a class effect of the coxibs. It was suggested that COX-2 selectivity resulted in an imbalance between prostacyclin and thromboxane production, an effect which would not be seen with traditional NSAIDs which inhibited the synthesis of both equally. Subsequent data from the prospective MEDAL6 and TARGET trials have not supported a class effect based on COX-2 selectivity. These studies suggest that treatment with either a coxib or an NSAID results in a small increase in cardiovascular risk. The risk is dose-related and rofecoxib, particularly at doses exceeding 50 mg per day, confers the highest cardiovascular risk in the majority of studies. A recent study of more than 1 million patients quantified cardiovascular risk as a composite of coronary death, non-fatal myocardial infarction and fatal and non-fatal stroke, and reported that diclofenac and rofecoxib were associated with the highest cardiovascular risk, while naproxen and perhaps celecoxib at doses ≤ 200 mg per day were the least likely to cause a cardiovascular event.7 NICE guidelines recommend that patients with pro-thrombotic risk, coronary artery or cerebrovascular disease should not be prescribed NSAIDs or a coxib. For other patients, treatment decisions should be made on an individual patient basis taking into account both cardiovascular and gastrointestinal risk factors. The medication should be prescribed for the shortest possible time and regularly reviewed.
Principal interactions
• Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers: there is a risk of renal impairment and hyperkalaemia.
• Quinolone antimicrobials: convulsions may occur if NSAIDs are co-administered.
• Anticoagulant (warfarin) and antiplatelet agents (dypiridamole, clopidogrel): increased risk of gastrointestinal bleeding with NSAIDs.
• Antihypertensives: their effect is lessened due to sodium retention by inhibition of renal prostaglandin formation.
• Ciclosporin and tacrolimus: nephrotoxic effect is exacerbated by NSAIDs.
• Cytotoxics: renal tubular excretion of methotrexate is reduced by competition with NSAIDs, with risk of methotrexate toxicity (low-dose methotrexate given weekly avoids this hazard).
• Diuretics: NSAIDs cause sodium retention and reduce diuretic efficacy and there is a risk of hyperkalaemia with potassium-sparing diuretics.
• Lithium: NSAIDs delay the excretion of lithium by the kidney and may cause lithium toxicity.
Paracetamol (acetaminophen)
Mode of action and uses
Paracetamol is an effective treatment for mild-moderate pain and for relieving fever. It does not affect platelet function or disrupt the GI mucosal barrier. Paracetamol has analgesic efficacy equivalent to aspirin, but in therapeutic doses it has only weak anti-inflammatory effects, a functional separation that reflects its differential inhibition of enzymes responsible for prostaglandin synthesis.8 For this reason, some would not class paracetamol as an NSAID.
Acute overdose
Severe hepatocellular damage and renal tubular necrosis can result from taking 150 mg/kg body-weight (about 10 or 20 tablets) in one dose.9 Patients at particular risk include:
• Those whose enzymes are induced as a result of taking drugs or alcohol; their liver and kidneys form more NAPQI.
• Those who are malnourished (chronic alcohol abuse, eating disorders, HIV infection) to the extent that the liver and kidneys are depleted of glutathione to conjugate with NAPQI.
Aspirin (acetylsalicylic acid)
Mode of action
• An antiplatelet effect due to permanent inactivation, by acetylation, of COX-1 in platelets, preventing synthesis of thromboxane A2. Platelets cannot regenerate the enzyme and the resumption of thromboxane A2 production is dependent on the entry of new platelets into the circulation (platelet lifespan is 7 days). Thus a continuous antiplatelet effect is achieved with low doses.
• Respiratory stimulation is a characteristic of aspirin intoxication and occurs both directly by stimulation of the respiratory centre and indirectly through increased carbon dioxide production.
• Although aspirin in high dose reduces renal tubular reabsorption of uric acid so increasing its elimination, other treatments for hyperuricaemia are preferred. Indeed aspirin should be avoided in gout as low doses inhibit uric acid secretion and on balance its effects on uric acid elimination are adverse.



