Chapter 29 Drugs and haemostasis
• Coagulation system: the mode of action of drugs that promote coagulation and that prevent it (anticoagulants) and their uses.
• Fibrinolytic system: the mode of action of drugs that promote fibrinolysis (fibrinolytics) and their uses to lyse arterial and venous thrombi (thrombolysis).
• Platelets: the ways that drugs that inhibit platelet activity benefit arterial disease.
The coagulation system
Coagulation initiates with tissue factor (TF), a cell membrane protein that binds activated factor VII (indicated by adding the letter ‘a’, i.e. factor VIIa). Although there is a small fraction of circulating factor VII in the activated state, it has little or no enzymatic activity until it is bound to TF. Most non-vascular cells express TF in a constitutive1 fashion, whereas de novo TF synthesis can be induced in monocytes and damaged endothelial cells. Injury to the arterial or venous wall exposes extravascular TF-expressing cells to blood. Lipid-laden macrophages in the core of atherosclerotic plaques are particularly rich in TF, thereby explaining the propensity for thrombus formation at sites of plaque disruption. Once bound to TF, factor VIIa activates factor IX and factor X (to IXa and Xa, respectively), leading to thrombin generation and clot formation (Fig. 29.1).
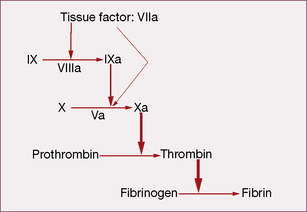
In the current model, blood coagulation starts with a transient release of tissue factor by damaged endothelium, resulting in the formation of sub-nanomolar amounts of thrombin via TF/VIIa-driven Xa formation (extrinsic-tenase). The initial thrombin activity is necessary to prime the system for a full thrombin explosion. Tissue factor pathway inhibitor (TFPI) rapidly shuts down this priming pathway and the full thrombin explosion is then dependent on factor IXa-driven Xa formation. Factor IXa-driven Xa formation (intrinsic-tenase) is amplified by the thrombin explosion itself, as thrombin forms a positive feedback loop by activating factor XIa (not shown in Fig. 29.1), which converts more IX to IXa.
• feedback activation of factor V and factor VIII
• activating platelet-bound factor XI, thereby leading to further factor Xa generation
• activating cells that provide the phospholipid surface required for assembly of the macromolecular enzymatic complexes.
Procoagulant drugs
Vitamin K
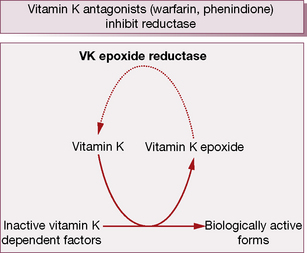
During γ-carboxylation of the proteins, the reduced and active form of vitamin KH2 converts to an epoxide, an oxidation product. Subsequently vitamin K epoxide reductase converts oxidised vitamin K back to the active vitamin K, i.e. there exists an interconversion cycle between vitamin K epoxide and reduced vitamin K (Fig. 29.2).
Vitamin K deficiency may arise from:
• bile failing to enter the intestine, e.g. obstructive jaundice or biliary fistula
• malabsorption syndromes, e.g. coeliac disease, or after extensive small intestinal resection
• reduced alimentary tract flora, e.g. in newborn infants and rarely after broad-spectrum antibiotics.
The following preparations of vitamin K are available:
Vitamin K is used to treat the following:
• Haemorrhage or threatened bleeding due to the coumarin or indanedione anticoagulants. Phytomenadione is preferred for its more rapid action; dosage regimens vary according to the degree of urgency and the original indication for anticoagulation.
• Haemorrhagic disease of the newborn, which develops usually between 2 and 7 days, and late haemorrhagic disease that presents at 6–7 months. Prophylaxis is recommended during the period of vulnerability with vitamin K (phytomenadione, as Konakion) 1 mg by single i.m. injection at birth. Alternatively, give vitamin K by mouth as two doses of a colloidal (mixed micelle) preparation of phytomenadione in the first week. Breast-fed babies should receive a further 2 mg at 1 month of age. Formula-fed babies do not need this last supplement as the formula contains vitamin K. Fears that intramuscular vitamin K might cause childhood cancer have been dispelled.
• Intestinal malabsorption syndromes; menadiol sodium phosphate should be used as it is water soluble.
Coagulation factor concentrates
Use of coagulation factor concentrates
• Superficial haemorrhage sometimes responds to local pressure.
• Minor bleeding can arrest with plasma factor concentrations of 0.25–0.30 units/mL, but severe bleeding requires at least 0.50 units/mL and surgical procedures or life-threatening haemorrhage require 0.75–1.00 units/mL by infusion of factor concentrate.
• In haemophilia A, factor VIII concentrate (t½ 8–12 h) is used for bleeding that is more than minor. Repeat dosing is necessary to maintain haemostatic levels.
• Factor IX (t½ 18–24 h) is used for bleeding that is more than minor in haemophilia B (Christmas disease).
• The speed of recovery of the affected joint or resolution of a haematoma determines the duration of therapy. After surgery, 7–14 days of replacement therapy is required to ensure adequate wound healing and to prevent secondary haemorrhage.
• Primary prophylaxis with factor concentrates two or three times weekly at doses sufficient to keep the factor above 0.01–0.02 units/mL reduces bleeding and hence the severity of chronic haemophilic arthropathy.
Desmopressin (DDAVP)
DDAVP is useful for treating patients with mild haemophilia A and von Willebrand’s disease, especially for short-term therapy. For dental extraction, a single injection of 0.3 micrograms/kg 1–2 h before surgery, combined with the oral antifibrinolytic drug, tranexamic acid, for 5–7 days after the procedure (see Antifibrinolytic drugs, p. 492), will produce normal haemostasis and prevent secondary haemorrhage.
DDAVP shortens the bleeding time in patients with renal or liver failure.
Anticoagulant drugs
• limiting thrombin generation, either as a result of inhibiting other proteases (clotting factors) involved in its generation or by reducing the activity of zymogens (the precursor inactive forms of the enzymes); or
• inhibiting (neutralising) thrombin activity, either directly or indirectly, depending on whether or not they activate the natural serpin-dependent anticoagulant pathway.2
Oral vitamin K antagonists (VKA)
Warfarin and other oral vitamin K antagonists (VKA) reduce the activity of zymogens.
During the γ-carboxylation of factors II (prothrombin), VII, IX and X (and also the natural anticoagulant proteins C and S), active vitamin K (KH2) is oxidised to an epoxide and must be reduced by the enzymes vitamin K epoxide reductase and vitamin K reductase to become active again (see the vitamin K cycle, p. 483). Coumarins3 are structurally similar to vitamin K and competitively inhibit vitamin K epoxide reductase and vitamin K reductase, so limiting availability of the active reduced form of the vitamin to form coagulant (and anticoagulant) proteins. The overall result is a shift in haemostatic balance in favour of anticoagulation because of the accumulation of clotting proteins with absent or decreased γ-carboxylation sites (PIVKAs).4
• maintain as stable a level of anticoagulation as possible
• adopt the lowest effective target INR
• educate patients about risk, particularly that associated with additional drug use.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


