drug toxicity and adverse effects, which discusses mechanisms for adverse effects produced both during normal drug therapy and after an overdose,

Drug toxicity and adverse effects
There is no consistent use of terminology in describing effects of drugs that were not intended when the drug was prescribed. Adverse effects of a drug are those that can occur when the drug is used at therapeutic doses; these have also been referred to as collateral effects. Drug toxicity is more commonly applied to effects that occur at supratherapeutic doses. This section provides a framework for classifying adverse and toxic effects of drugs which are referred to in the individual drug monographs as unwanted effects. The term side effect is a widely used alternative.
The beneficial effects of a drug in one situation (e.g. the antidiarrhoeal effect of opioids) may be an adverse effect in other circumstances (e.g. constipation, when an opioid is used for pain relief). Therefore, even classification of the nature of effect into beneficial or adverse may depend on the condition being treated. The adverse effects caused by different drugs are listed in the British National Formulary (BNF), and it is apparent that for most drugs’ potential adverse effects are more numerous than beneficial properties. It is important to remember that such lists are not exhaustive, and prescribers should be alert to both predicted and unexpected reactions to medicines. The prescriber should consider the risk–benefit ratio for each individual and the suitability of alternative drugs and/or treatments and people who are prescribed drugs should be informed of the risk–benefit balance inherent in their treatment. The patient information leaflet (PIL) included with the dispensed medicine represents a useful way of providing such advice. Prescribers should also be aware of the risks of adverse and toxic effects arising from drug interactions.
It should be appreciated that all drugs are associated with some risk of adverse effects and toxicity, although both the severity and incidence differ widely among drugs. In general, the acceptability of a risk of an unwanted effect is related to the severity of the disease being treated; for example, serious idiosyncratic reactions with incidences of 1 in 10 000 have led to the withdrawal of some non-steroidal anti-inflammatory drugs (NSAIDs), whereas some cancer chemotherapeutic agents can cause significant toxicity in nearly all individuals.
A useful indication of the safety margin available for a drug is given by the therapeutic index (TI):
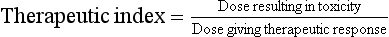
Drugs such as diazepam have a TI of about 50 and it is difficult for even the most inept doctor to cause serious toxicity with diazepam. In contrast, digoxin has a TI of only about 2, and for such drugs toxicity may be precipitated by relatively small changes in dosage regimen or the clearance of the drug from the body. The TI relates to serious toxicity and does not indicate the potential for minor adverse effects, which may inconvenience the person enough for him or her to stop treatment but are not considered to represent drug toxicity.
Types of unwanted effect
Adverse drug reactions are usually divided into two main types:
 Type A (Augmented): these effects are usually dose-related and largely predictable from the known pharmacological and biochemical effects of the drug or its metabolites,
Type A (Augmented): these effects are usually dose-related and largely predictable from the known pharmacological and biochemical effects of the drug or its metabolites,
This classification makes no allowance for time-dependency of some adverse events, or for the underlying susceptibility of the individual. Other types of adverse reactions have been defined as: continuing (type C), delayed (type D), end-of-use (type E), failure of therapy (type F) or genetic/genomic (type G). These types are not mutually exclusive, and classification of an adverse event along multiple axes, such as dose-relatedness, time-relatedness and susceptibility may be more meaningful.
Pharmacological toxicity
In ‘pharmacological toxicity’, the toxic reaction is a predictable extension of the known pharmacology of the drug at its site(s) of action (Table 53.1), and should be recognised readily when monitoring the individual’s response to treatment. There are numerous examples in this book where the adverse effect is really an excessive therapeutic action.
Table 53.1
Examples of drugs with adverse effects related to their primary therapeutic properties
| Drug | Adverse effect |
| Acetylcholinesterase inhibitors | Muscle weakness |
| Beta-adrenoceptor antagonists | Heart block when used as an antiarrhythmic |
| Insulin | Hypoglycaemia |
| General anaesthetics | Medullary depression |
| Loop diuretics | Hypokalaemia |
| Warfarin | Haemorrhage |
For many drug effects, the response increases with increase in dose, with low sub-therapeutic doses giving an inadequate response, therapeutic doses giving the desired response, but very high doses giving an excessive response that can be regarded as a form of toxicity (response 1 in Fig. 53.1). A good example is warfarin (Ch. 11), inadequate doses of which are associated with a lack of the desired anti-thrombotic effect, whereas at excessive doses there is a risk of haemorrhage. This has given rise to the concept of a ‘therapeutic window’, which is a range of doses or blood/plasma concentrations within which most individuals should show a beneficial response with minimal risk of adverse effects (illustrated by response 1 in Fig. 53.1). The concept is particularly valuable in the interpretation of measurements of drug concentrations in plasma, which can be used to monitor adherence to treatment and to assess likely response (Table 53.2).
Table 53.2
Examples of therapeutic windows based on plasma concentrations
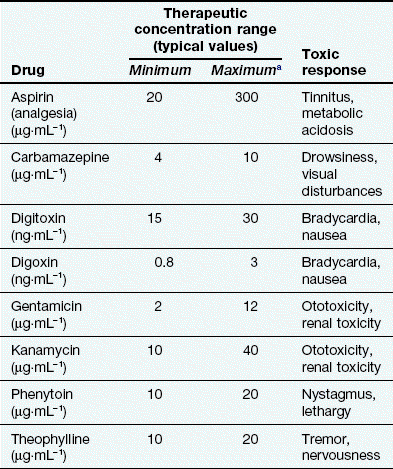
aThe maximum concentration may be limited by toxicity related to the primary therapeutic response (e.g. carbamazepine) or to an unrelated effect (e.g. gentamicin).
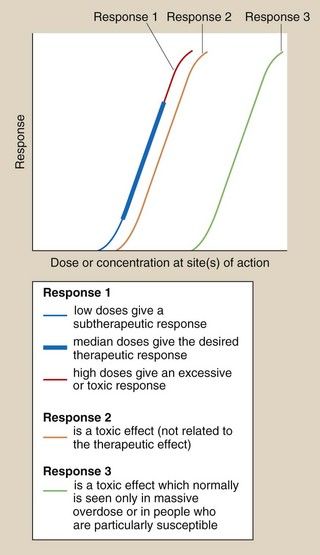
Fig. 53.1 Dose–response relationships in relation to toxicity.
Response 1 is the primary therapeutic effect, the magnitude of which increases with dose from sub-therapeutic, through therapeutic to potentially toxic. Response 2 is an undesired effect seen at a dose only slightly greater than those producing the therapeutic effect. Response 3 is an adverse effect normally seen only in overdose.
In many other cases, the toxic reaction may be unrelated to the primary therapeutic effect (Table 53.3), and may be caused by a secondary effect that is not the primary aim of the treatment given (response 2 in Fig. 53.1). This toxicity will often be present to a limited extent at appropriate therapeutic doses.
Table 53.3
Examples of drugs with adverse effects unrelated to their primary therapeutic use
| Drug | Adverse effects |
| Beta-adrenoceptor agonists | Increase in heart rate when used in asthma |
| Beta-adrenoceptor antagonists | Reduction in heart rate when used for hypertension |
| Aminoglycosides | Deafness |
| Anti-cancer drugs | Myelosuppression |
| Anticonvulsants | Sedation when used for epilepsy |
| Antipsychotics | Dystonias or parkinsonism |
| Corticosteroids | Glaucoma |
| Drugs for Parkinson’s disease | Hallucinations and confusion |
| Opioid analgesics | Respiratory depression |
| Paracetamol | Liver failure |
| Statins | Rhabdomyolysis |
| Thalidomide | Birth defects |
| Thiazides | Glucose intolerance |
The separation of therapeutic and toxic dose–response curves is a measure of the TI. If these are very close (e.g. responses 1 and 2 in Fig. 53.1), then there is a low safety margin (low TI) and most individuals will show some degree of toxicity at normal therapeutic doses, for example myelosuppression with cytotoxic anti-cancer drugs. For drugs with widely separated therapeutic and toxic dose–response curves (e.g. responses 1 and 3 in Fig. 53.1) – that is, those with high TIs – toxicity would not be seen at normal doses; for example a β-adrenoceptor antagonist would be very unlikely to cause myocardial depression and heart failure in people with normal left ventricular function. However, the toxic effect may occur in individuals who are unusually sensitive because of their genetics or their physical condition; for example, standard doses of a β-adrenoceptor antagonist can precipitate heart failure in people with pre-existing impaired left ventricular function.
Pharmacological toxicity is the most common cause of adverse effects. Such toxicity can be minimised by an assessment of the risk–benefit balance for the individual to be treated. This should take into account factors that may influence both pharmacokinetics and target-organ sensitivity, including age, physiological status (e.g. renal function), concurrent medication, disease processes, environmental factors (e.g. smoking), etc.
Because of the predictable nature of pharmacological toxicity, it is usual for some treatments to be co-prescribed with drugs that will reduce the possibility of toxic effects; examples include anti-emetics given with cancer chemotherapy, vitamin B6 given with isoniazid and leucovorin (folinic acid) given after high-dose methotrexate.
Biochemical toxicity
In ‘biochemical toxicity’, the toxicity or tissue damage is caused by an interaction of the drug, or an active metabolite, with cell components, especially macromolecules such as structural proteins and enzymes. A generalised scheme is given in Figure 53.2. For most licensed drugs this form of toxicity is identified and characterised during preclinical studies in animals and monitored in clinical trials (Ch. 3), for example by measuring changes in serum levels of liver or muscle enzymes.
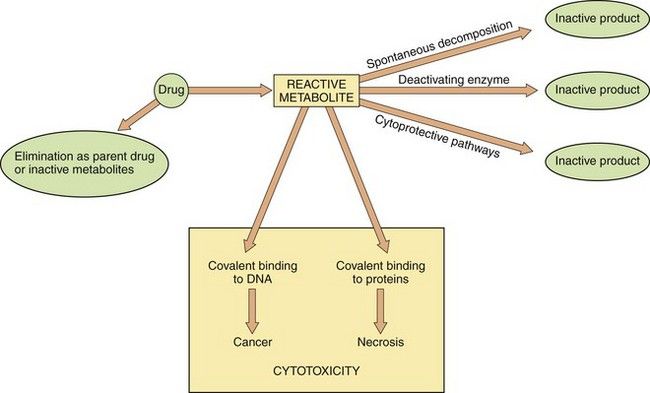
Fig. 53.2 Metabolism and cytotoxicity.
In this scheme, the propensity of the reactive metabolite of a cytotoxic drug to cause adverse biochemical effects on cellular DNA and proteins depends on the balance between formation of the reactive metabolite from the parent drug, and the rate of elimination of both the reactive metabolite and the parent drug by alternative metabolic routes. Therapeutic interventions are aimed at either increasing elimination of the parent drug or enhancing cytoprotective pathways to reduce the cytotoxic effects.
In some situations, an understanding of the mechanism of toxicity has allowed the development of appropriate treatments or antidotes. An example is the key observation that the thiol (-SH) group of the endogenous tripeptide glutathione can prevent cell damage caused by highly reactive chemical species, such as the toxic metabolite of paracetamol (see below and Fig. 53.3). The nature of the cell damage is related to the stability of the toxic reactive chemical; extremely unstable metabolites may bind covalently to and inactivate the enzyme that forms them, whereas more stable species may be able to diffuse to a distant site, for example DNA, and initiate changes such as cancer. Important examples of biochemical toxicity are given below.
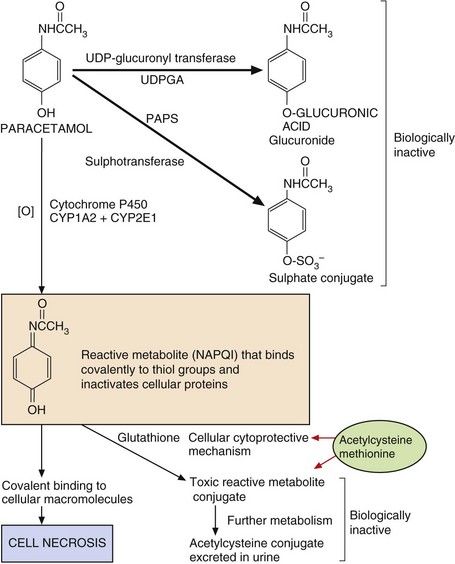
Fig. 53.3 Pathways of paracetamol metabolism.
In overdose, the concentrations of 3′-phosphoadenosine 5′-phosphosulphate (PAPS) (for sulphation) and glutathione (for cytoprotection) are depleted, and extensive macromolecular binding leads to hepatocellular necrosis. N-Acetylcysteine and methionine replenish glutathione to conjugate the toxic metabolite. NAPQI, N-acetyl-p-benzoquinone imine; UDPGA, uridine diphosphate glucuronic acid.
Paracetamol
Paracetamol-induced hepatotoxicity represents the result of an imbalance between metabolic detoxication of paracetamol, via conjugation with glucuronic acid and sulphate, and metabolic activation to an unstable toxic metabolite (N-acetyl-p-benzoquinone imine, NAPQI) via oxidation by cytochrome P450. This toxic metabolite can bind covalently to proteins and cause cell necrosis. Low doses of paracetamol are safe because they are eliminated mainly by conjugation, and any NAPQI created by cytochrome P450 oxidation is inactivated by a cytoprotective pathway involving glutathione. However, in overdose, the sulphate conjugation reaction is saturated and there is increased cytochrome P450-mediated oxidation of paracetamol to NAPQI (Fig. 53.3). Once the hepatic glutathione is depleted this toxic metabolite binds covalently to proteins and causes oxidative stress and cell necrosis. This biochemical mechanism explains:
 the site of toxicity (centrilobular necrosis in the liver because of the large amounts of cytochrome P450 present),
the site of toxicity (centrilobular necrosis in the liver because of the large amounts of cytochrome P450 present), the increased toxicity seen in individuals treated with inducers of cytochrome P450 (especially alcohol-related induction of CYP2E1),
the increased toxicity seen in individuals treated with inducers of cytochrome P450 (especially alcohol-related induction of CYP2E1), the increased toxicity seen in individuals with low hepatic stores of glutathione due to poor nutrition,
the increased toxicity seen in individuals with low hepatic stores of glutathione due to poor nutrition,
Cyclophosphamide
Cyclophosphamide is an anti-cancer drug. Its highly toxic metabolites bind to DNA as part of their mechanism of action, but they are eliminated in the urine and cause haemorrhagic cystitis due to cell damage in the bladder (Ch. 52). This adverse effect can be prevented by prior treatment with mesna (mercaptoethane sulphonic acid), which possesses both a thiol group for cytoprotection and a highly polar sulphonic acid group, and results in high renal excretion and delivery of this cytoprotective molecule to the bladder epithelium. It is usually given either orally 2 h before the cyclophosphamide or intravenously at the same time to cover the period of maximum urinary excretion of toxic cyclophosphamide metabolites. It is not yet known whether mesna also protects against bladder cancer, which can arise about 10–20 years after initial treatment with cyclophosphamide.
Isoniazid
Isoniazid, which is used for the treatment of tuberculosis (Ch. 51), causes hepatitis in about 0.5% of treated individuals. This is believed to result from the formation of a reactive metabolite, N-acetylhydrazine, which is produced by acetylation followed by oxidative metabolism. The biochemical basis for the susceptibility of some individuals to the hepatotoxic metabolite is not known. Fast acetylators (see Ch. 2) form more N-acetylhydrazine than do slow acetylators, but, unexpectedly, they are not more sensitive to isoniazid toxicity. Susceptibility may be related to the balance between further activation of N-acetylhydrazine (by oxidation) and its detoxification (by further acetylation); if this is so, then fast acetylators may produce more active metabolite but also inactivate it more rapidly.
Spironolactone
Spironolactone (Ch. 14) is oxidised by cytochrome P450. The metabolite formed in the testes binds to and destroys testicular cytochrome P450 and this causes a decrease in the metabolism of progesterone to testosterone (a step which is also catalysed by a cytochrome P450). This effect, combined with an anti-androgenic action at receptor sites (pharmacological toxicity), can result in gynaecomastia and decreased libido.
Aromatic amines and nitrites
Aromatic amines, such as the anti-leprosy drug dapsone and some antimalarials (e.g. primaquine), are oxidised in the liver to active metabolites, which are released into the circulation, where they can affect erythrocytes, causing methaemoglobinaemia and/or haemolysis.
Methaemoglobinaemia: In the erythrocyte, the active metabolite interacts with molecular oxygen (O2), which then oxidises haemoglobin (Fe2+) to methaemoglobin (Fe3+) and also oxidises the active metabolite itself (Fig. 53.4A). Because of the large amounts of haemoglobin compared with the amount of drug given this would be inconsequential, were it not for the fact that the oxidised active metabolite can be recycled back to the active metabolite by reduction with NADPH (reduced nicotinamide adenine dinucleotide phosphate) in the erythrocyte. Consequently, each molecule of the metabolite undergoes repeated redox cycling and is able to oxidise many molecules of haemoglobin. NADPH is formed during the metabolism of glucose 6-phosphate by glucose-6-phosphate dehydrogenase (G6PD) (Fig. 53.4A). The activity of G6PD, and hence the amounts of NADPH, are determined genetically. There is a high incidence of G6PD deficiency in people of African ancestry and very high in those of Mediterranean ancestry, such as Kurdish people. Such individuals have limited NADPH reserves and a low ability to reduce the oxidised active drug metabolite (Fig. 53.4A) back to the active metabolite. In consequence, there is limited redox cycling of the active drug metabolite and they are less susceptible to drug-induced methaemoglobinaemia, but they are more susceptible to haemolysis (see below).
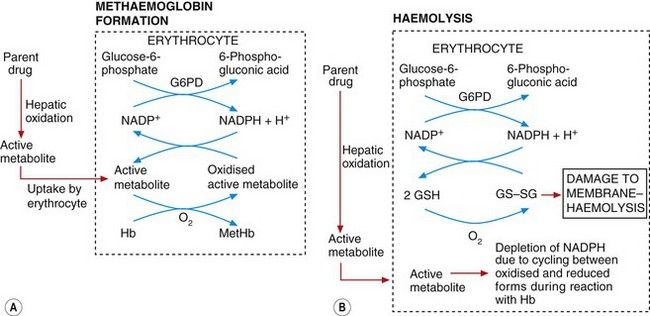
Fig. 53.4 Mechanisms of methaemoglobinaemia and haemolysis.
In methaemoglobinamia (A), the active drug metabolite oxidises haemoglobin (Hb) to methaemoglobin (MetHb). The oxidised active metabolite is repeatedly recycled to the active metabolite by reduced nicotinamide adenine dinucleotide phosphate (NADPH) generated by glucose-6-phosphate dehydrogenase (G6PD). In (B), the depletion of NADPH as a result of methaemoglobinaemia depletes levels of the reduced glutathione (GSH) necessary for maintaining erythrocyte cell membrane integrity; the build-up of oxidised glutathione dimers (GS–SG) causes membrane damage and haemolysis. The active metabolite may also react directly with glutathione to lower GSH concentrations.
Haemolysis: This arises from an increase in erythrocyte membrane permeability associated with accumulation of oxidised glutathione (GS–SG in Fig. 53.4B) in the erythrocyte. Oxidised glutathione accumulates because redox cycling of the drug metabolite linked to the formation of methaemoglobin (Fig. 53.4A) (see above) results in depletion of NADPH, which is the cofactor essential for maintaining glutathione in the reduced state. Individuals with G6PD deficiency are very susceptible to haemolysis caused by aromatic amines and nitrites because the low endogenous levels of NADPH are depleted rapidly and oxidised glutathione cannot then be reduced. Given the geographical distribution of G6PD deficiency, it is ironic that the amino groups associated with this form of toxicity are often present in drugs used to treat tropical infections (such as primaquine for the treatment of malaria; see Ch. 51 and Box 47.6).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


