Fig. 56.1 Sales of thalidomide and the incidence of phocomelia.
Thalidomide sales and phocomelia incidence are each expressed as a percentage of the reported maximum.
The list of drugs known to be teratogenic is relatively short but includes thalidomide, many anticonvulsants, some chemotherapeutic drugs (e.g. alkylating agents and antimetabolites), warfarin, androgens, danazol, diethylstilbestrol, lithium and retinoids (Table 56.1). Because of their long half-lives, some retinoids can result in teratogenesis even if treatment for the mother is stopped before pregnancy occurs. Although teratogenesis is commonly thought of in terms of structural abnormalities or dysfunctional growth in utero, it also refers to long-term functional defects. For example, maternal consumption of alcohol during pregnancy may cause behavioural and cognitive abnormalities in childhood, despite the birth of a seemingly unaffected infant. Some drugs may initially appear harmless yet exhibit a long latency period. Diethylstilbestrol, which was given during pregnancy between the 1940s and early 1970s in the mistaken belief that it reduced the risk of miscarriage, resulted in abnormalities in the offspring when they reached adulthood, including hypogonadism in males and vaginal adenocarcinoma in females.
Table 56.1
Examples of drug-induced teratogenicity and fetal/neonatal toxicity
| Therapeutic drug | Teratogenic and adverse effects in fetus and neonate |
| ACE inhibitors | Affect fetal and neonatal blood pressure control and renal function; oligohydramnios |
| Alcohol | Fetal alcohol syndrome; growth restriction (Ch. 54) |
| Aminoglycosides | Auditory or vestibular nerve damage |
| Amiodarone | Neonatal goitre |
| Androgens | Virilisation of female fetus |
| Anti-cancer drugs | Carcinogenic and teratogenic effects (also avoid before pregnancy) |
| Barbiturates | Fetal abnormalities; withdrawal effects in neonates |
| Benzodiazepines | Withdrawal effects in neonates |
| Beta-adrenoceptor antagonists | Intra-uterine growth restriction, neonatal hypoglycaemia and bradycardia |
| Carbamazepine | Neural tube defects |
| Carbimazole | Neonatal goitre |
| Corticosteroids | Intra-uterine growth suppression (with prolonged treatment) |
| Dapsone | Neonatal haemolysis and methaemoglobinaemia |
| Diethylstilbestrol | Hypogonadism in male offspring and vaginal cancer in female offspring |
| Fibrinolytics | Premature separation of placenta in first 18 weeks |
| Lamotrigine | Teratogenicity |
| Leflunomide | Teratogenic in animals; effective contraception necessary for at least 2 years after end of treatment for women and 3 months for men |
| Lithium salts | Teratogenicity; cardiac abnormalities |
| NSAIDs | Premature closure of ductus arteriosus; pulmonary hypertension |
| Opioids | Neonatal respiratory depression and risk of withdrawal syndrome if the mother is habituated |
| Oral anticoagulants | Malformations; fetal or neonatal haemorrhage |
| Oxcarbazepine | Neural tube defects |
| Phenytoin | Congenital malformations; risk of neonatal haemorrhage due to vitamin K deficiency |
| Primaquine | Neonatal haemolysis and methaemoglobinaemia |
| Retinoids and retinoid-like drugs | Teratogenic, craniofacial malformations; some have long half-lives and effective contraception is essential for prolonged periods after stopping treatment and before pregnancy |
| Ribavirin | Teratogenic in animals; effective contraception necessary for at least 6 months after treatment for both women and men |
| Statins | Decreased cholesterol synthesis affects fetal development |
| Sulphonamides | Neonatal haemolysis and methaemoglobinaemia |
| Sulfonylureas | Neonatal hypoglycaemia |
| Thiazide diuretics | Growth retardation; electrolyte disturbance |
| Valproate | Congenital malformations and developmental delay in offspring |
Manufacturers of most drugs advise that they should be taken in pregnancy only if the potential benefit outweighs the possible risk; also, many recommend that prescribing to women of childbearing age should be carried out with pregnancy in mind and contraception should be adequate before, during and after treatment. ACE, angiotensin-converting enzyme; NSAIDs, non-steroidal anti-inflammatory drugs.
Notwithstanding the limited list of drugs that are known to cause teratogenesis, there is a much larger number that should be avoided or used with caution in pregnancy because of their potential to produce detrimental effects in the fetus. Examples include warfarin-induced anticoagulation (Ch. 11), which may predispose to cerebral haemorrhage in the fetus during delivery; in contrast, heparin is an effective anticoagulant in the mother and does not cross the placenta. Non-steroidal anti-inflammatory drugs (NSAIDs; Ch. 29) can produce premature closure of the ductus arteriosus before delivery. Adverse effects produced at therapeutic doses, such as tachycardia with tricyclic antidepressants and growth restriction with corticosteroids, may also affect the fetus or neonate.
Whenever a drug is given to a pregnant woman, or a woman who has child-bearing potential, an assessment should be made, taking into account any risk to the fetus balanced against the benefit to the mother and any risk associated with not treating the mother. For example, treatment with anticonvulsants or antimalarials may be essential for the mother, despite the possible risk to the fetus/neonate. The risk to the fetus/neonate should be minimised whenever possible by selecting the drug with the least potential for teratogenicity, by prescribing the lowest effective dose and minimizing the use of multiple drugs. Absence of evidence of teratogenicity does not imply that a risk does not exist. Drugs which have been used extensively in pregnancy and appear to be safe should nevertheless be preferred to new or untried drugs. The BNF provides detailed information on potential adverse drug effects on the fetus and neonate, and guidance is also available from the UK Teratology Information Service (www.uktis.org).
Pharmacokinetics in pregnancy
The placenta provides a potential barrier to the transfer of macromolecules from the maternal circulation, but low-molecular-weight drugs will cross the placenta, particularly if they are lipid-soluble. Some metabolism of drugs can occur in the placenta, which may further restrict fetal exposure, although the placenta does not have a high drug-metabolising capacity. The fetal liver and kidneys have only modest abilities to eliminate drugs, so drugs in the fetal circulation are usually cleared by the maternal routes of elimination. The fetus therefore represents a slowly equilibrating maternal kinetic compartment, with transfer across the placenta being determined by the concentration gradient between fetal and maternal circulations.
Maternal pharmacokinetics are affected by a number of physiological changes, especially in late pregnancy. Compared to non-pregnant women, these include:



These changes mean that maternal drug concentrations are often lower than those in a non-pregnant woman given the same dose, so drug doses may need to be increased in pregnancy to compensate.
Drugs and breastfeeding
Almost any compound present in the maternal circulation will enter breast milk and be ingested by the suckling baby. However, with a few exceptions there is little evidence that drug intake via breastfeeding is of concern because most drugs enter breast milk in quantities too small to affect the baby. In general, drugs licensed for use in children can be safely given to the nursing mother, whereas drugs known to have serious toxic effects in adults, or known to affect lactation, such as bromocriptine, should be avoided.
The American Academy of Pediatrics (2001) divides drugs into:
The reader should refer to the up-to-date information in the BNF for detailed advice. The absence of safety information for many drugs in lactation means that only essential drugs should be given to mothers during breastfeeding.
Pharmacokinetics in lactation
Several factors influence drug transfer from the maternal circulation into breast milk, including the characteristics of the milk, the physicochemical properties of the drug and the amount of drug in the maternal circulation. The concentrations of drugs in breast milk are in equilibrium with those in the maternal circulation. At equilibrium, the free concentration in milk and plasma will be the same, but the total concentration will be influenced by the extent of protein binding and uptake into the lipid phase (see Fig. 2.3). Water-soluble drugs diffuse from plasma into milk, and the concentration in breast milk is similar to the non-protein-bound fraction in the maternal plasma. Lipid-soluble compounds diffuse into breast milk and may concentrate because of the high fat content in milk.
The effects of drugs in breast milk depend not only on maternal pharmacokinetics but also on the extent of absorption, distribution and elimination of the drug in the neonate or infant (see below). Drugs may also have different pharmacodynamic properties in neonates or infants compared to older children and adults. If drugs are given during breast feeding, compounds with short half-lives are preferred because they are less likely to accumulate in neonates, who have lower drug clearance. Neonatal exposure can also be minimised if the feed is timed to coincide with the trough blood concentration in the mother, which is just before a dose is taken. The World Health Organization (WHO) nevertheless recommends that the health and developmental benefits of breastfeeding are usually greater than any likely risk from drugs in breast milk.
Prescribing for children
Both the pharmacokinetics and responses to drugs may differ in neonates, infants and children compared with adults. There are considerable differences among neonates (<1 month), infants (1–12 months) and children, because many metabolic and physiological processes are immature at birth and develop rapidly in the first months of life. These differences may affect the absorption and distribution of drugs and the rate of elimination of the drug from the body, and also the sensitivity of tissues to its actions or adverse effects. Particular care is needed in prescribing drugs that may affect growing or maturing organ systems such as the bones and teeth and the reproductive system. Box 56.1 shows some of the differences between the young and adults.
Box 56.1 Developmental changes in the young that may alter drug handling compared with adults
Low production of gastric acid and erratic gastric emptying in first year of life
Smaller gut surface area/body mass ratio, but greater gut permeability to larger molecules
Greater proportion of body fat and larger extracellular volume may alter the volumes of distribution of some drugs
Maturation of drug metabolising enzyme pathways in the liver occurs at different rates over the first year
Glomerular filtration rate and tubular secretion are relatively low in the first year of life
Although medicines should usually be used within the terms of the product license, many drugs given to children have not undergone formal clinical evaluation in this age group, and are not specifically licensed for paediatric use. It is recognised that ‘off-label’ prescribing may be necessary and the UK Medicines Act (1968) does not prohibit such unlicensed use. There is an increasing recognition of the need for formal clinical trials in the paediatric population, but such studies raise significant ethical issues. The BNF for Children (www.bnf.org/bnf/index.htm) gives specific guidance on prescribing for children in the UK and further information on the regulation of paediatric medicines is available at the following places.
Pharmacokinetics in neonates and children
In neonates, inefficient metabolism and renal clearance mean that lower doses of some drugs are needed after allowing for body weight, and doses need to be calculated with special care. The processes of drug elimination are largely mature by a few weeks of age, after which drug clearance (adjusted to body weight) is similar to or higher than that in adults (see below). However, children may be more susceptible to effects on growing or maturing tissues and organs. Generalisations are difficult and each drug needs to be considered in its own right.
Absorption
Slow rates of gastric emptying and intestinal transit may reduce the rate of drug absorption in neonates, but total absorption of poorly absorbed drugs may eventually be more complete because of longer contact with the intestinal mucosa. In the neonate, gastric pH is neutral and this can reduce the absorption of weak acids but increase the absorption of weak bases.
Distribution
Neonates and young children have a lower body fat content and higher total body water than adults; this influences the distribution of both lipid- and water-soluble drugs. Neonates have a lower plasma albumin concentration, which also has a lower affinity for drug binding. In addition, the higher plasma concentrations of free fatty acids and bilirubin compete with drugs for plasma protein-binding sites. The overall effect is reduced plasma protein binding, which not only increases the apparent volume of distribution of the drug but also increases the proportion of drug able to cross the blood–brain barrier; it also increases the amounts diffusing into the liver and therefore available for metabolism. Drugs that are strongly bound to albumin should not be used during neonatal jaundice because the drugs may displace bilirubin (which is mostly in the unconjugated form) from protein-binding sites and increase the risk of kernicterus.
Metabolism
The drug-metabolising enzyme systems are immature in the neonatal liver, and first-pass metabolism and hepatic drug clearance are low, especially for substrates of CYP1A2, CYP3A4 and glucuronidation. The clearances for substrates for these enzymes are two to six times lower in neonates compared with adults. When the enzyme systems mature, drug metabolism processes become more extensive. Plasma drug clearance is often higher in young children than in adults, because of their higher relative liver mass and greater hepatic blood flow per kilogram of body weight; hepatic blood flow is the rate-limiting step in the elimination of high-clearance drugs.
Renal elimination
Renal function in the neonate and infant is much less developed than in children or adults. The glomerular filtration rate in the newborn is about 40% of the adult level, and tubular secretory processes are poorly developed. Elimination of drugs such as digoxin, gentamicin and penicillin will therefore be slower until about 6–8 months of age.
In children, the larger volume of distribution and faster hepatic elimination mean that doses of metabolised drugs need to be higher than in adults after correcting for the difference in body weight. Prescribed doses are most accurately judged by considering both age and body surface area. In children, body surface area is a better guide to appropriate drug dosage than body weight. It can be estimated from a nomogram or by using the Du Bois formula:
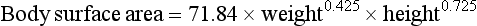
where body surface area is in square metres, weight in kilograms and height in metres. The drug dose for a child can be then approximated as:
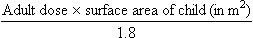
where 1.8 m2 is the average body surface area of a 70 kg adult.
Prescribing for the elderly
The elderly (usually taken to mean those over 70 years old) comprise a heterogeneous group who show considerable variation in ‘biological’ age. Changes occur in both the pharmacodynamics and pharmacokinetics of drugs with increasing age.
The density or numbers of receptors may be reduced with age; for example, β-adrenoceptors decrease in number, reducing the response to agonist drugs. The elderly are often more susceptible to sedatives, hypnotics and antipsychotic drugs, possibly because of changes in receptor numbers and/or reduced efficiency of the blood–brain barrier. They are also more susceptible to the adverse effects of NSAIDs on the gut.
Altered structure and function of target organs can also influence the effects of drugs. For example, baroreceptor function is impaired in the elderly and vasodilator drugs are more likely to provoke postural hypotension. The high peripheral resistance and less distensible arterial tree found with increasing age also respond less well to arterial vasodilators.
These changes reflect the ageing process itself, but they are often complicated by the presence of chronic disease (frequently involving multiple pathological processes) and variation due to both genetic and environmental influences. The risks of unwanted effects are higher in the elderly as a consequence of these changes. Significant numbers of hospital admissions in the elderly are due to adverse drug reactions, most of which are the more predictable type A effects (see Ch. 53). In addition, drug interactions are more common in the elderly because of the coexistence of different treatable conditions requiring the simultaneous use of several drugs (polypharmacy). For these reasons, it is usual to start drug treatment in the elderly with the smallest effective dose. Rational prescribers should also seek to minimise the numbers of drugs used, with clear explanations of usage instructions and regular review of drug regimens.
Pharmacokinetics in the elderly
Drug absorption across the gut wall is not greatly affected by ageing, although bioavailability may be increased due to reduced first-pass metabolism.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


