Drug Interactions
OBJECTIVES
The reader will be able to:
- Define the following terms: additivity, antagonism, displacer, combination products, competitive inhibition, drug interaction, inhibition constant, isobologram, mechanism-based inhibition, noncompetitive inhibition, synergism, therapeutic drug interaction.
- Discuss the graded nature of drug interactions, and provide two examples involving reversible inhibition, two involving mechanism-based inhibition, and one involving induction.
- Explain mechanistically and kinetically the difference between reversible and mechanism-based inhibition.
- Ascertain whether pharmacokinetics or pharmacodynamics of a drug, or both, is altered by another drug, given response and unbound plasma drug concentration–time data following chronic administration.
- Anticipate the likely changes in plasma and unbound concentrations with time when the pharmacokinetics of a drug is altered by concurrent drug administration.
- Explain why displacement of plasma protein binding is unlikely to produce a clinically significant drug interaction.
- Explain why the degree of interaction tends to be greater when the affected drug is administered orally than when administered parenterally, for drugs that are normally subjected to a high oral first-pass metabolic loss.
- Explain why the risk of a severe interaction resulting from inhibition is greater in poor than extensive metabolizers, when the fraction of drug metabolized polymorphically in extensive metabolizers is high, and the inhibitor inhibits elimination via the other pathway(s).
- Anticipate the likely changes in plasma and unbound concentrations with time when the pharmacokinetics of a drug is altered by concurrent drug administration.
- Show graphically the consequence of a pharmacokinetic drug interaction when the mechanism and the circumstances of its occurrence are given.
- Explain why the combination of two agonists or two antagonists of the same receptor may not produce a greater response than that achieved with any one of them.
 atients commonly receive two or more drugs concurrently; indeed, inpatients on average receive five drugs during a hospitalization, and many, particularly elderly, patients may be taking concurrently eight or more drugs, as discussed in Chapter 14, Age, Weight, and Gender, Fig. 14-4. In addition, with an increasingly aged population, the issue of polypharmacy is of ever greater concern. The reasons for multiple-drug therapy are many. One reason, as discussed in Chapter 9, Therapeutic Window, is that drug combinations have been found to be beneficial in the treatment of some conditions, including various cardiovascular diseases, infections, and cancer. Another reason is that patients frequently suffer from several concurrent diseases or conditions, and each may require the use of one or more drugs, in which case the number of drug combinations is huge. Furthermore, drugs may be prescribed by clinicians in different medical specialties each of whom may be not fully aware of the therapeutic maneuvers of the others. Finally, patients may also selfmedicate with over-the-counter medicines or herbal preparations. All of these reasons lead to a high probability of a drug interaction during drug therapy, especially in the elderly.
atients commonly receive two or more drugs concurrently; indeed, inpatients on average receive five drugs during a hospitalization, and many, particularly elderly, patients may be taking concurrently eight or more drugs, as discussed in Chapter 14, Age, Weight, and Gender, Fig. 14-4. In addition, with an increasingly aged population, the issue of polypharmacy is of ever greater concern. The reasons for multiple-drug therapy are many. One reason, as discussed in Chapter 9, Therapeutic Window, is that drug combinations have been found to be beneficial in the treatment of some conditions, including various cardiovascular diseases, infections, and cancer. Another reason is that patients frequently suffer from several concurrent diseases or conditions, and each may require the use of one or more drugs, in which case the number of drug combinations is huge. Furthermore, drugs may be prescribed by clinicians in different medical specialties each of whom may be not fully aware of the therapeutic maneuvers of the others. Finally, patients may also selfmedicate with over-the-counter medicines or herbal preparations. All of these reasons lead to a high probability of a drug interaction during drug therapy, especially in the elderly.
A drug interaction occurs when either the pharmacokinetics or pharmacodynamics of one drug is altered by another. In addition, constituents of some foods and herbal preparations may alter the pharmacokinetics or pharmacodynamics of drugs. Examples are inhibition by grapefruit juice of gut wall CYP3A and induction of this enzyme by St. John’s wort, discussed in Chapter 7, Absorption, and Chapter 12, Variability. As the principles are the same as for drugs, such food and herbal drug interactions are not considered further, although they can be therapeutically important.
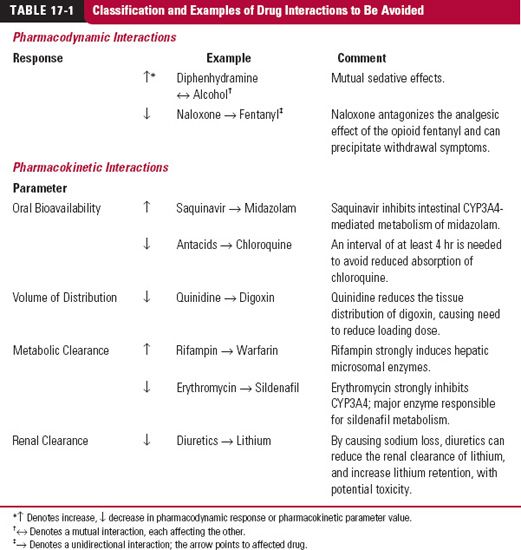
The potential for interactions among drugs within the body are almost limitless and are a source of variability in drug response. Many regard drug interactions as all-or-none, viewing them either as occurring or not, whereas in reality, most drug interactions, whether pharmacokinetic or pharmacodynamic in nature, are graded, depending on the concentration of the interacting drugs, and hence on the dosage regimens and pharmacokinetics of the drugs. Few of these interactions are of a type or of a sufficient magnitude to be clinically important. However, a number are and the risk of them occurring increases with the number of drugs that the patient is taking. A therapeutic drug interaction has then occurred. It is prudent to provide a cautionary note for those situations in which the two (or more) drugs need to be coprescribed. Some examples are listed in Table 17-1; in most cases, the interaction results in an exaggerated response of the affected drug, but not always so. Reduced activity can arise because the perpetrating drug either is an antagonist, reduces bioavailability, or increases clearance. Occasionally, the magnitude of the interaction is so great as to contraindicate the simultaneous use of the drugs. Very occasionally, the interaction has been so much more severe than anticipated and the likelihood of the combination being inadvertently given so great to cause either the affected or offending drug to be removed from the market. Examples are the antihistamines terfenadine and astemizole, the antihypertensive mebifredril, the statin cerivastatin, and cisapride, used to treat heartburn. However, the drugs were not removed from the market until some major clinical adverse events, sometimes fatal, occurred. Inadvertent, and potentially avoidable, adverse drug interactions also comprise a significant source of hospital admissions.
Not all patients receiving several interacting drugs elicit a therapeutically significant interaction. Often, it is just a few. The reasons for these differences in response among patients are manifold. Included are individual differences in the dosage regimen and duration of administration of each drug, in the sequence of drug administration, and in patient adherence. Pharmacodynamic and pharmacokinetic differences in genetics, concurrent disease states, and many other factors also contribute. Thus, the circumstances associated with a clinically significant interaction in an individual should always be carefully documented.
A pharmacodynamic interaction can arise for various reasons. These include the interacting drug complementing the action of the other, such as the use of a thiazide diuretic and a β-blocker, each acting by a different mechanism to lower blood pressure. Alternatively, the interacting drug may act as an antagonist or agonist at the same receptor site as the other drug, and, if an agonist, it may act additively or synergistically. The effect is additive if the combined effect is that expected based on the concentration–response curves for each drug given independently. Synergism occurs when the effect produced with the two drugs is even greater than expected had the effect been additive.
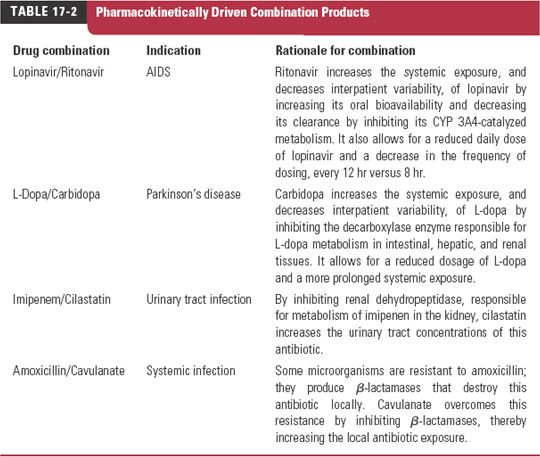
In the case of pharmacodynamic interactions, for which the mechanism of action of each compound is known, the interaction is generally predictable and, if undesirable, is avoidable. The same is increasingly true of pharmacokinetic interactions, and when the interaction is desirable, the interacting drugs are often prescribed as combinations of fixed doses, although in such cases, there is commonly only one active principle; the second compound interacts to overcome an existing limitation in one or more aspects of the pharmacokinetics of the affected drug to increase its exposure–time profile at the target site for a given dose. Examples of pharmacokinetically driven beneficial combination products are listed in Table 17-2. Detailed aspects of some of these are discussed later in this chapter. Furthermore, most attention clinically in the area of drug interactions has been directed to changes in systemic exposure of drugs, and this forms the major emphasis of the current chapter.
Before considering drug interactions in greater detail, two final general comments need to be made. The first concerns the sequence of drug administration. A drug interaction is most likely to be detected in clinical practice when the interacting drug is initiated or withdrawn. For example, given the usual variability in patients’ responses to drugs, it is unlikely that a drug interaction would be detected if the affected drug is titrated to response in a patient already stabilized on the drug causing the interaction. Certainly, the dosage regimen of the affected drug would be different in that patient than would otherwise be the case, but the resulting regimen may still be within the normal range. In this case, only if the offending drug is withdrawn first, when the patient is stabilized on the drug combination, would the interaction be seen. The interaction would also have been detected if the interacting drug had been administered to the patient already stabilized on the original drug. However, sometimes the delay in full expression of the interaction may take so long that the clinician may not associate a change in patient response to the addition or withdrawal of the offending drug.
The second final comment concerns interactions involving drug transporters. Recall from Chapter 7, Absorption, that drugs that belong to class 1 of the Biopharmaceutics Classification System are highly soluble and permeable (e.g., Table 7-9); their absorption, and most of their disposition, is unaffected by transporters. The reason is that passive permeability tends to predominate. For analogous reasons drug interactions involving inhibition or induction of transporters, certainly in relation to intestinal absorption and probably hepatic processes, are not anticipated to affect the pharmacokinetics of such drugs. A clear potential exception exists at the blood-brain barrier, because there, owing to a low passive permeability, even for class 1 compounds, distribution may be permeability rate-limited. Inhibition of transporters there could result in altered brain permeability, and hence changes in both rate and extent of brain distribution, even of class 1 compounds. However, examples to date are rare. Class 1 drugs can nonetheless be inhibitors or inducers of transporters, and so potentially may affect the pharmacokinetics of drugs in classes 2 to 4 of the Biopharmaceutics Classification System.
CLASSIFICATION
One system of classifying drug interactions is to note whether drug response is increased or decreased. While perhaps clinically useful, this classification does not help to define the mechanism of the interaction. In this book, interactions are classified based on whether pharmacokinetics or pharmacodynamics is altered; occasionally, both are changed. Distinction between the two is made by relating response to the unbound plasma concentration of the pharmacologically active species. No change in the unbound concentration–response curve implies a pharmacokinetic drug interaction, which can arise either through a chemical interaction, such as inhibition of metabolism, or through a physiologic process, such as altered blood flow at an absorption site. The result is a change in one, or more, of the primary pharmacokinetic parameters, ka, F, V, CLR, CLH, that in turn alters the secondary pharmacokinetic parameters, such as half-life and fe.
Before proceeding, a discussion of what is meant by the word interaction is worthwhile. Strictly speaking, this word implies a mutual effect. The interaction between two drugs, A and B, might thus be denoted by A ↔ B. An example is the competition between two drugs for a common binding site on albumin, one drug displaces but is also displaced by the other. Generally, however, the term interaction is interpreted more broadly to indicate any situation in which one drug affects another. For example, phenobarbital appears to reduce the absorption of the diuretic, furosemide, but the renal clearance of phenobarbital is increased by the diuresis produced by furosemide. This might be regarded as a bidirectional interaction and may be denoted by A  B. Clearly, in the case of mutual and bidirectional interactions, the measured response of one drug cannot be considered without also defining the level of the other.
B. Clearly, in the case of mutual and bidirectional interactions, the measured response of one drug cannot be considered without also defining the level of the other.
When given in sufficient quantities, two drugs almost always affect each other; this may not be the case, however, at concentrations achieved in therapy. For example, the antibiotic enoxacin inhibits the metabolism of theophylline, but theophylline, at doses normally given, does not affect the response or the pharmacokinetics of enoxacin. This interaction is unidirectional and may be denoted by A → B. In a unidirectional interaction, the unaffected Drug A (e.g., enoxacin) can be considered independently, but the change in response of the affected Drug B (e.g., theophylline) cannot be adequately defined without also considering the concentration–response curve of the effect of Drug A on Drug B. Examples of different types of drug interactions are listed in Table 17-1.
Finally, various terminologies have been used to describe the drugs involved in an interaction. The one producing the interaction has been called the offending drug, the precipitant, the perpetrator, or the villain (when the outcome is adverse). The one affected has been called the affected drug, the object, or the victim. In this chapter, these terms are used interchangeably.
Emphasis in this chapter is on pharmacokinetic drug interactions, although some aspects of pharmacodynamic interactions are considered toward the end. The physiologic concepts developed in Chapter 4, Membranes and Distribution, Chapter 5, Elmination, and Chapter 7, Absorption, together with the kinetic principles developed in Chapter 9, Therapeutic Window, Chapter 10, Constant-Rate Input, and Chapter 11, Multiple-Dose Regimens, form the basis for discussing many aspects of pharmacokinetic drug interactions. For convenience, in this chapter, the effect of one drug on another is examined under the separate headings of altered absorption, altered distribution, and altered clearance. However, it should be borne in mind that several pharmacokinetic parameters can be, and sometimes are, altered simultaneously, examples of which are subsequently discussed.
ALTERED ABSORPTION
It was stated in Chapter 6, Kinetics Following an Extravascular Dose, and Chapter 11, Multiple-Dose Regimens, that the more rapid the absorption process, the higher and earlier is the peak plasma concentration and that neither total area under the curve (AUC) after a single dose nor AUC within a dosing interval at plateau after chronic dosing changes unless the bioavailability of the drug is altered. Therapeutic consequences of a change in either rate or extent of drug absorption were discussed in Chapter 11, Multiple-Dose Regimens.
Considered now are situations in which bioavailability is altered, with emphasis on changes in average plasma concentration with time during chronic dosing; an altered speed of absorption (ka) changes only the degree of fluctuation around the average value. The events with respect to a constant rate of drug input are illustrated in Fig. 17-1.
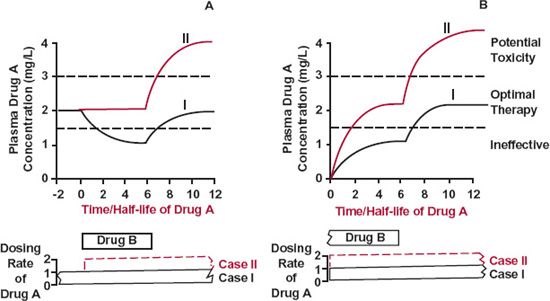
FIGURE 17-1. Altered absorption. As long as Drug B is administered, the bioavailability of Drug A is reduced by one half. For simplicity, Drug A is assumed to be given at a constant rate. The therapeutic concentration range of Drug A lies between 1.5 and 3 mg/L. A. Patient stabilized on Drug A. The plasma concentration of Drug A falls by one half (Case I), unless the dosing rate is doubled (Case II, color), when Drug B is concurrently administered. A problem potentially arises if administration of Drug A is not reduced to its preexisting rate when Drug B is removed (Case II, color). B. Patient stabilized on Drug B. An adequate concentration of Drug A is not achieved (Case I), unless the usual dosing rate of this drug is doubled (Case II, color). When Drug B is withdrawn, there is a potential problem similar to that considered in A. In both A and B, the time course of events for Drug A is determined by its half-life.
Figure 17-1A depicts the situation in which a patient stabilized on Drug A, now receives Drug B, which reduces the bioavailability of Drug A. It is apparent that if no steps are taken to change the dosing rate of Drug A, its concentration falls to a lower plateau, the time being determined by the elimination half-life of this drug. Suppose that it is then noticed that the patient is no longer being treated effectively with Drug A, and that, having recognized the problem, the offending drug is withdrawn. The concentration of Drug A would then return to the previous plateau value in 3.3 elimination half-lives. Therapeutic control would again be restored. Alternatively, on another occasion, in anticipation of the problem but desiring to give the two drugs together, the dosing rate of Drug A is increased appropriately at the time that Drug B is introduced. As long as Drug B continues to be administered, therapeutic control is satisfactory. A problem arises, however, if Drug B is subsequently withdrawn, but the dosing rate of Drug A is not correspondingly reduced. Then, in 3.3 elimination half-lives, the concentration of Drug A reaches a higher plateau where toxicity is likely.
Another possible situation is one in which Drug A is added to the regimen of a patient stabilized on Drug B (Fig. 17-1B). Here, as before, optimal therapy with Drug A in the presence of Drug B is achieved only if the usual dosing rate of Drug A is appropriately increased. However, the danger of the concentration of Drug A rising too high, if the dosing rate of Drug A is not readjusted when Drug B is withdrawn, must always be kept in mind.
The situations just considered emphasize the dependence of time course of events on the half-life of the affected drug. Thus, for affected drugs with very long half-lives, such as phenobarbital (4-day half-life), changes in response are insidious, occurring over several weeks, and the clinician may not associate the interaction with the causative drug, which was either initiated or stopped quite some time previously.
ALTERED DISTRIBUTION
One drug can affect the kinetics of tissue distribution of another, for example, by changing tissue perfusion, a hemodynamic interaction. This occurs during general anesthesia when perfusion to many tissues is reduced. However, primary consideration here is directed to changes in the extent of distribution, reflected in changes in volume of distribution, V. Recall the relationship for drugs of V≥ 50 L (≥0.7 L/kg)

from which it is seen that V can be altered by changes in either fu or fuT, the fraction unbound in that tissue space. Although theoretically possible, changes in VP, the volume of plasma, and VT, the volume of aqueous space of tissues into which drug distributes, are extremely unlikely to occur. Recall also when V <50 L, and particularly for very low volume of distribution drugs (V <15 L or <0.20 L/kg), we need to take into account that changes in fu give rise to corresponding changes in binding to the same plasma protein residing in the interstitial spaces. Another cause of an altered distribution, considered later, is one in which a drug inhibits the uptake or efflux by a transporter of another drug whose distribution is permeability rate-limited.
The most common explanation for altered distribution in a drug interaction is displacement. Displacement is the reduction in the binding of a drug to a macromolecule, usually a protein, caused by competition of another drug, the displacer, for common binding site(s). The result is a rise in fu or fuT, or in both. Sometimes, binding is diminished through an allosteric effect. The second drug, binding at another site, induces a conformational change in the protein, thereby reducing the affinity of the first drug for the protein. Occasionally, an allosteric effect causes an enhanced affinity between drug and protein; drug binding is then increased.
CONDITIONS FAVORING DISPLACEMENT
Two conditions must be met before substantial displacement occurs. First, in the absence of a displacer, the drug must be bound mostly to a protein, that is, its fu or fuT must be low. Obviously, if a drug is not bound, it cannot be displaced. Second, the displacer must occupy most of the binding sites, thereby substantially lowering the number of sites available to bind drug.
Table 17-3 lists two groups of acidic drugs that bind to albumin. As can be seen, not all drugs bind to, and hence compete for, the same primary binding site on albumin. Albumin and some other proteins have several binding sites, each exhibiting some degree of specificity. Hence possessing an acidic function is not in itself a sufficient criterion for predicting the ability of one acidic drug to displace another. Moreover, even though almost all those drugs that compete for the same site have a high affinity for albumin, only a few are generally listed clinically as displacers, such as salicylic acid, valproic acid, and ibuprofen. This list is limited because the bound plasma concentration achieved during therapy must approach or exceed 0.6 mM (140 g/3 L), the molar concentration of plasma albumin. For a substance with a molecular weight of 250, this concentration corresponds to 150 mg/L, but even a concentration 20% of this value, 30 mg/L, may increase fu significantly. These concentrations are approached during salicylate and valproic acid therapy and during high-dose ibuprofen therapy, because these drugs are given in doses approaching 1 g, they are of low molecular weight (138, 144, and 204 g/mol, respectively) and they possess relatively small volumes of distribution, 10 to 15 L/70 kg. High molar concentrations could also be achieved with lower daily doses if the compound has a long half-life relative to dosing interval, thereby resulting in extensive accumulation. For example, approximately 40% of a 25-mg daily dose of leuflonamide is converted to a very highly albumin-bound active metabolite (molecular weight = 270 g/mol), which, because of its very low clearance (0.01 L/hr) and long half-life (~10 days), yields plateau plasma metabolite concentrations in the order of 50 mg/L.
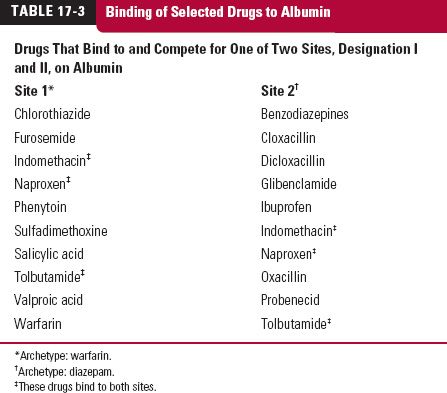
Displacers share an expected common property (i.e., their fu values change with plasma concentration). With most sites occupied by the displacer, the fraction unoccupied is sensitive to a change in the concentration of displacer. Therefore, displacers must show concentration-dependent disposition kinetics (Chapter 16, Nonlinearities).
So far, distinction has been made between displacer and displaced drug, but it should be apparent that this classification is arbitrary. Valproic acid is said to displace warfarin from albumin, but only because the therapeutic plasma concentration of valproic acid (70 mg/L; 0.48 mM) approaches the molar concentration of albumin (0.6 mM), whereas that of warfarin (1–4 mg/L; 0.003–0.01 mM) does not. Both drugs have high affinity for the same site on albumin, and if the concentrations were reversed, warfarin would be called the displacer.
The conclusions drawn from the interactions between acidic drugs and albumin are generally applicable to all drug–protein interactions, bearing in mind the widely differing molar concentrations of the various binding proteins in plasma (Chapter 4, Membranes and Distribution). For example, the molar concentration of α1-acid glycoprotein, which avidly binds many basic drugs, is low (0.009–0.023 mM), and therefore displacement interactions can occur at plasma concentrations much lower than those required for displacement of acidic drugs from albumin (600-mM concentration). Substantial displacement of bases from tissue binding, observed as a decrease in V, is less likely, however, because unlike in plasma, where α1-acid glycoprotein is the primary binding protein, there are relatively high concentrations of the major binding proteins, the acidic phospholipids, in tissues. Or, stated differently, the unbound tissue concentrations of many bases are well below the Kd of the binding constituents.
THERAPEUTIC IMPLICATIONS
Displacement interactions in plasma have been studied primarily in vitro. The potential displacer is added at therapeutic concentrations to a sample of plasma containing the drug, and changes in its binding are measured. Substantial displacement is frequently demonstrated, yet this may be of little therapeutic consequence. Much depends on whether the events are acute or occur at plateau during chronic therapy.
Acute Events. Two situations can be envisaged. One involves the administration of a loading dose of drug to a patient already stabilized on a displacer. The other involves the administration of a dose of displacer to a patient already stabilized on a drug. In both situations, the question of altering the usual dose of drug arises only if the unbound drug concentration increases above the therapeutic range, in the presence of the displacer. The extreme situation is one in which the displacer is administered as an intravenous (i.v.) bolus, which initially is restricted essentially to plasma, causing a transient rise in the unbound displaced drug there, before falling back as both displacer and unbound displaced drug moves down their respective concentration gradients into tissues. Accordingly, if response tracks unbound plasma concentration more rapidly than the kinetics of redistribution, a transient increase in response might be anticipated. In practice, this last situation is unlikely to arise, because to minimize the risk of adverse reactions, most intravenously administered drugs are given as a short-term infusion rather than as a bolus.
Less transient events are expected if, after redistribution, the unbound volume of distribution (Vu) of displaced drug decreased significantly, because this will produce a sustained increase in unbound drug for a given amount in the body. Such a decrease in Vu occurs only if, in the absence of the displacer, the drug is substantially bound in the body and the displacer causes significant displacement of drug from the major binding sites. For example, the unbound concentration rises if displacement occurs from sites on a plasma protein for a drug with a small volume of distribution, around 10 L/70 kg, or if displacement occurs from tissue-binding sites for a drug with a large volume of distribution. An example of the former situation is the displacement of warfarin from albuminbinding sites by naproxen. Displacement from tissue-binding sites would be expected to occur with high doses of bosentan and imirestat coadministered with drugs that bind to the same sites, given that these sites are saturated at such high doses (Chapter 16, Nonlinearities). However, these situations are relatively uncommon. More common is displacement from plasma-binding sites of a drug with a large volume of distribution. Then, because so little drug resides in plasma, relative to that in the body as a whole, only a minimal change in the unbound drug concentration, and hence response, occurs even when all the drug on the proteins are displaced, as discussed in Chapter 4, Membranes and Distribution. For example, if the volume of distribution is 100 L and 99% of the drug in plasma is bound (fu = 0.01), then only 3% (100 × [1 − fu]) · ([plasma volume]/V) of that in the body resides bound in plasma. Even if completely displaced, the small amount affected would redistribute throughout the rest of the body and so would only marginally increase the unbound body pool.
Returning to the example of warfarin, because the clinical response (prothrombin time) is so delayed and insensitive to acute changes in warfarin concentration (Fig. 8-7, Chapter 8, Response Following a Single Dose), no adjustment in dosage of this oral anticoagulant would be contemplated in the event of acute displacement, even though Vu is reduced. A more important consideration for most other drugs is the effect of displacement on events at plateau, because most drugs, including displacers, are given chronically.
Events at Plateau. The influence of displacement on the unbound concentration at steady state (Cuss) depends on extraction ratio and route of administration of the affected drug.
Consider a drug with a low extraction ratio. Recall that clearance depends on fu, but unbound clearance (CLu) does not. Also recall the equalities (Eq. 5-2, Chapter 5, Elimination)
and that for this class of drug, CLu approximates CLint. When drug is infused at a constant rate, Rinf, and steady state is achieved,
Rinf = CLint · Cuss = CL · Css 17-3
However, because intrinsic clearance is unaffected by displacement, the same is true of the value of Cuss. Thus, although displacement by increasing fu increases clearance (and hence causes the total plasma concentration to fall), no change in response and therefore in dosing rate is anticipated at steady state. Neither is any change anticipated in Cuav,ss upon chronic multiple dosing.
Now consider a drug with a high extraction ratio. Because blood clearance is unaffected by displacement, the steady-state blood concentration is also unaffected following a constant-rate i.v. infusion. However, as binding is diminished, (fuT↑), Cuss and therefore response to the drug must be increased as Cuss = fub · Cb,ss. The maintenance dosing rate of the drug may need to be reduced. Although an extremely uncommon situation, conceivably the opioid fentanyl, which has a high extraction ratio, is reasonably highly bound (fu = 0.15–0.2) and administered both intravenously and transdermally, fulfills the condition, but no clinically significant displacement interaction has been reported.
Prediction of the outcome when a drug with a high extraction ratio is given orally and when elimination occurs predominantly in the liver is difficult. In the presence of the displacer, both unbound drug clearance and oral bioavailability decrease (see Chapter 7, Absorption); the effect of displacement on the unbound drug concentration at plateau is therefore likely to be relatively small. Irrespective of any effect of displacement, however, the unbound concentration at plateau is lower than that achieved following an equivalent i.v. infusion.
The events predicted at plateau before and after displacement for drugs of low and high extraction are shown in Fig. 17-2. Shown in Fig. 17-3 are the total and unbound plateau plasma concentrations of the antiepileptic drug phenytoin in patients before and after receiving valproic acid, another antiepileptic drug, which displaces phenytoin from albumin binding sites. As predicted for a drug of low extraction, the unbound concentration is unaltered, whereas the total plasma concentration is decreased. Accordingly, based on these observations, there is no need to alter the dosing rate of phenytoin in patients receiving valproic acid.
Kinetic Features. Both acute events and events at plateau have been discussed. Remaining to be discussed are the kinetic consequences of displacement on approach to plateau under the usual condition in which both drug and displacer are given chronically. To do so, first consider the expected changes in disposition kinetics of a drug upon displacement.
Disposition Kinetics. The subsequent points summarize those made in Chapter 5, Elimination. Because clearance of a drug with a high extraction ratio is unaffected by displacement, the half-life changes with volume of distribution. In contrast, for a drug of low extraction, because unbound clearance, CLu (CLint,) does not change, half-life either remains constant (when Vu does not change) or shortens (if Vu decreases) with displacement from plasma proteins.
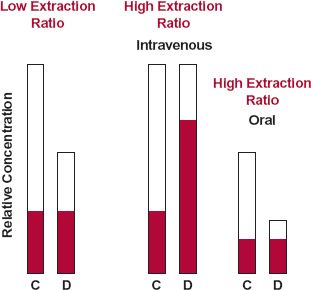
FIGURE 17-2. Changes, at plateau, in the total ( and
and  ) and unbound (
) and unbound ( ) concentrations (D = displaced; C = control) depend on the extraction ratio of the drug and route of drug administration. For a drug of low extraction ratio, diminished binding (fu ↑) reduces the total, but not the unbound, concentration regardless of the route of administration. For a drug of high hepatic extraction ratio, clearance, and so total concentration, is unaffected by diminished binding when the drug is given intravenously. The unbound concentration, however, is elevated. Because diminished binding decreases oral bioavailability of the highly cleared drug, the likely outcome of displacement after oral administration is a decrease in the total concentration and little or no change in the unbound concentration. The actual change depends on whether the effect on oral bioavailability or clearance predominates.
) concentrations (D = displaced; C = control) depend on the extraction ratio of the drug and route of drug administration. For a drug of low extraction ratio, diminished binding (fu ↑) reduces the total, but not the unbound, concentration regardless of the route of administration. For a drug of high hepatic extraction ratio, clearance, and so total concentration, is unaffected by diminished binding when the drug is given intravenously. The unbound concentration, however, is elevated. Because diminished binding decreases oral bioavailability of the highly cleared drug, the likely outcome of displacement after oral administration is a decrease in the total concentration and little or no change in the unbound concentration. The actual change depends on whether the effect on oral bioavailability or clearance predominates.
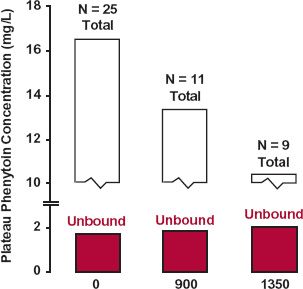
FIGURE 17-3. The valproic acid–phenytoin interaction involves displacement only. Although plasma protein binding of phenytoin is decreased when sodium valproate is administered chronically to a group of patients stabilized on phenytoin, with a resultant fall in the steady-state plasma phenytoin concentration, there was no substantial change in the unbound phenytoin concentration (colored). These observations are consistent with a displacement interaction of phenytoin by valproic acid. Note that the degree of phenytoin displacement depends on the dose of sodium valproate. Of the 25 patients stabilized on phenytoin, 11 received 900-mg sodium valproate per day; 9 received a 1350-mg daily dose; and some received both regimens. (From: Mattson RH, Cramer JA, Williamson PD, Novelly, RA. Valproic acid in epilepsy: clinical and pharmacological effects. Ann Neurol 1978;3:20–25.)
Plasma Concentration–Time Profile. Although there are many possible combinations of events, only one common situation is considered. In this case (Fig. 17-4), the displacer, which has a longer half-life than that of the drug, is added to the regimen of a patient stabilized on a low extraction ratio drug. To simplify matters, both drug and displacer are administered by constant-rate infusion and both are assumed to always be at distribution equilibrium. Although these conditions are somewhat restrictive, the situation chosen does illustrate the importance of both kinetics and the manner of drug administration on the likely therapeutic outcome on approach to plateau.
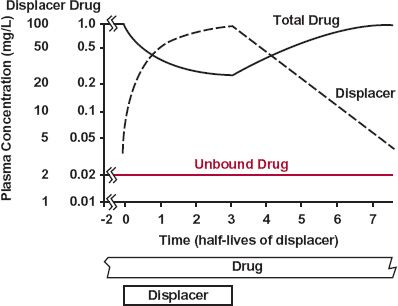
FIGURE 17-4. When constantly infused, the unbound concentration (color) of a drug with a low extraction ratio remains virtually unchanged if a displacer with a long half-life, relative to the drug, is either infused or withdrawn. The change in total plasma drug concentration reflects the displacement.
Notice in Fig. 17-4 that slow accumulation and correspondingly slow elimination of the displacer results in insignificant changes in the unbound drug concentration and therefore response. This is a consequence of the kinetics of the displacer being slower than that of the drug. The concentration of the displacer is changing so slowly relative to that of the drug that at all times the drug is at a virtual steady state, with rate of elimination (CLint · Cuss) matching the rate of input. That is, although there is a tendency for the unbound drug concentration to rise above the concentration at steady state during accumulation of the displacer, there is an opposing tendency for the unbound concentration to fall because the rate of elimination (CLint · Cu) would then exceed the rate of administration (CLint · Cuss). The reverse tendencies occur on stopping the displacer, but once again, they balance each other out. Accordingly, although the unbound concentration remains essentially unaltered, the total plasma drug concentration changes inversely with that of the displacer, reflecting the changing degree of displacement. A transient change in unbound concentration might occur if the kinetics of the drug, rather than that of the displacer, had been the slower.
In the case just discussed, no change in effect, and hence in the dosage regimen of the drug, is anticipated even though displacement has occurred. If only response were monitored, displacement would not have been suspected.
ALTERED CLEARANCE
A reduction in unbound clearance is potentially the most dangerous type of drug interaction. The unbound drug concentration can then rise to a toxic level, unless a reduction in dosage is made. Consequently, it is extremely important to be able to identify, characterize, and, where possible, avoid this type of adverse interaction. Interactions involving an increase in unbound clearance, because of induction, also occur causing the unbound drug concentration to fall, potentially resulting in a diminished response. Although generally of less concern, a diminished response can sometimes lead to failure of therapy with its attendant risks. Both inhibition and induction often have a high degree of specificity involving a specific enzyme. Examples of inhibitors and inducers of the major drugrelated CYP enzymes are illustrated in Fig. 17-5. Inhibition and induction of transporters also occurs, but currently far less is known about specific transporters than enzymes.
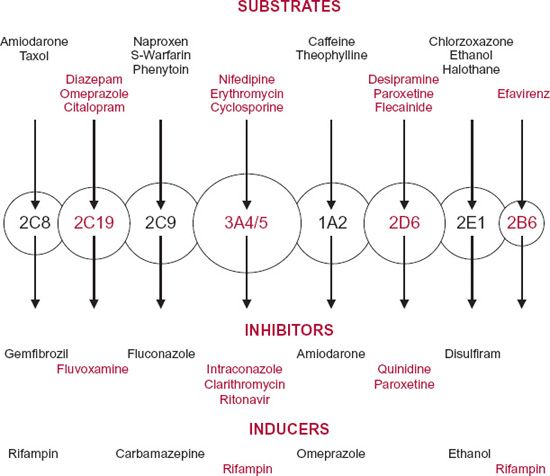
FIGURE 17-5. Graphic representation of the different forms of cytochrome-P450 (circles) in humans with different but some overlapping substrate specificities. The arrows indicate single metabolic pathways. Representative substrates are listed above for each enzyme. Also listed are relatively selective inhibitors and inducers of the enzymes.
INHIBITION
Inhibition of drug metabolism is the major cause of reduced unbound clearance. A reduction can also occur on inhibition of biliary and renal secretion and on inhibition of an uptake transporter, when uptake permeability rate-limits, or becomes the rate-limiting step in, organ clearance. Inhibition is of two general types, competitive and noncompetitive. Competitive inhibition occurs when inhibition can be overcome by raising sufficiently high the concentration of the substrate. It is the most common type of inhibition. The net effect is an increase in the apparent Km of the substrate but no change in its Vm. The term is normally applied to enzyme metabolism, but, more broadly, can be applied also to transport processes, with an associated increase in observed KT but no change in Tm. Noncompetitive inhibition, which although less common is no less important, involves independent inhibition of an enzyme and, unlike competitive inhibition, cannot be overcome by raising the concentration of the substrate; it is characterized by a decrease in Vm but no change in Km of the substrate. One common type of noncompetitive inhibition is mechanism-based inhibition involving the formation of a reactive metabolite that effectively covalently binds to, and permanently inactivates, the enzyme. We first focus on competitive inhibition and then mechanism-based inhibition.
Competitive Inhibition
A Case Study. Some patients receiving theophylline develop nausea when enoxacin, an anti-infective quinoline, is added to the theophylline regimen. The nausea is associated with an elevated concentration of theophylline caused by enoxacin inhibiting the metabolism of theophylline.
The dramatic rise in the trough plasma concentration of theophylline when enoxacin is added to the regimen, and the return to the pre-enoxacin value when enoxacin is withdrawn, are illustrated in Fig. 17-6. Notice that it takes approximately 4 days of enoxacin administration for theophylline concentration to rise to the new plateau and a similar time for it to return to the pre-enoxacin value after enoxacin is withdrawn.
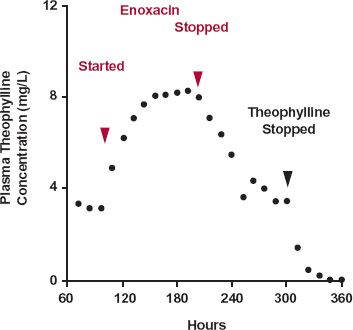
FIGURE 17-6. Enoxacin inhibits theophylline elimination. Subjects received 150-mg theophylline (in a controlled-release dosage form) orally every 12 hr for 12 days (288 hr). By 72 hr, as expected, the trough has reached a plateau with an average concentration within a dosing interval of 4.8 mg/L (not shown). Enoxacin was given, 400 mg orally every 12 hr for 4 days, starting 96 hr into the theophylline regimen. On addition of enoxacin, the mean trough theophylline concentration rose to a new and higher plateau (average interdose concentration of 9 mg/L, not shown
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


