Renal elimination involves the transfer or excretion of the parent drug from the blood to the renal tubule, from where it is subsequently eliminated in the urine. Metabolism, which takes place primarily in the liver, involves the conversion of the parent drug to another molecular species (metabolite) through the action of an enzyme. Metabolites may undergo further metabolism, renal excretion, and/or biliary excretion. It is important to note that when the parent drug is altered chemically and converted to a metabolite, the drug is considered to be eliminated, and that the subsequent fate of the metabolite is not part of the pharmacokinetic profile of the parent drug. If the metabolite possesses some therapeutic and/or toxic activity, a separate study of its pharmacokinetics would be of clinical importance.
5.1.1 First-Order Elimination
Renal excretion and metabolism are first-order processes for over 90% of all drugs. Elimination that is not first-order results in nonlinear pharmacokinetics, which is discussed in Chapter 15. As first-order processes, the rates of excretion and metabolism may be expressed
(5.1) 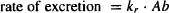
(5.2) 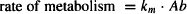
where kr and km are the first-order rate constants (units, time−1) for renal excretion and metabolism, respectively, and Ab is the amount of drug in the body.
The rate of overall elimination, which is the sum of all component processes, may be expressed
(5.3) 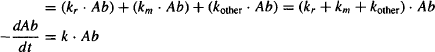
where kother is the sum of the first-order rate constants for any processes other than renal excretion or metabolism, and k, the sum of all the rate constants, is known as the overall elimination rate constant. Because most drugs are eliminated by renal excretion or metabolism, k ≈ kr + km.
Elimination of a drug can also be expressed in terms of its elimination half-life (t1/2) (see Appendix B):
(5.4) 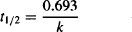
The half-life and the elimination rate constant are both measures of the speed with which a drug is eliminated. One is the reciprocal form of the other. Under normal circumstances they are constants for a drug and their values can be obtained from the literature. A drug that has a large elimination rate constant will have a short half-life and will be eliminated rapidly. Conversely, a drug that has a small elimination rate constant will have a long half-life and will be eliminated slowly. Clinically, the half-life is used more frequently because it has more meaning. For example, if a drug has a half-life of 7 h, it is clear that the plasma concentration and the amount of drug in the body will fall by half every 7 h. An elimination rate constant of 0.1 h−1 provides the same information but has much less direct practical meaning.
5.1.2 Determinants of the Elimination Rate Constant and the Half-Life
The half-life and the elimination rate constant are simply parameters of a first-order process. It is important to understand how they are related to physiological processes in the body and the disposition characteristics of a drug. The half-life and the elimination rate constant are known as secondary or derived parameters, because they are derived from, or determined by, two primary drug parameters: the primary parameter for elimination (clearance) and the primary parameter for distribution (volume of distribution).
Clearance is a measure of the efficiency with which the liver and/or kidney can extract a drug from the plasma and eliminate it from the body. High values of clearance will promote large elimination rate constants. However, for the liver and kidney to have the opportunity to eliminate a drug, the drug must be present in the plasma. As a result, a drug’s volume of distribution, which reflects the relative distribution of a drug between the plasma and the rest of the body, is also an important determinant of the elimination rate constant and half-life. If a drug has a large volume of distribution, a large fraction of the drug in the body will be located in the tissues and will not be accessible to the organs of elimination. This will impede the body’s efforts to remove the drug and will promote a long half-life.
- A large clearance and a small volume of distribution will promote rapid elimination and a short half-life.
- A small clearance and a large volume of distribution will promote slow elimination and a long half-life.
5.2 CLEARANCE
Clearance is the most important of all the pharmacokinetic parameters. As we show in Chapter 11, it controls the average plasma concentration achieved from a particular rate of drug administration. If a patient is suspected to have altered clearance for a drug, the dose may have to be adjusted appropriately or the drug may have to be avoided altogether.
5.2.1 Definition and Determinants of Clearance
Clearance is the primary or fundamental pharmacokinetic parameter for elimination. It expresses the collective ability of the organs of elimination to remove drug from plasma. Because drugs are eliminated primarily by renal excretion and/or hepatic metabolism, total clearance is made up primarily of renal clearance and hepatic clearance, which express the ability of the kidney and liver, respectively, to eliminate drug presented to them in the blood. A drug’s clearance is a constant under normal circumstances but can be altered by diseases of the liver and/or kidney, concomitant medications, and other factors that affect renal and/or hepatic function.
Clearance, by definition, is the constant of proportionality between the rate of elimination and the plasma concentration:
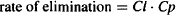
Although the processes of renal and hepatic clearance are very different, both elimination processes involve the extraction of drug from the blood as it flows through the organ. As a result, the efficiency of the elimination process is expressed in terms of the volume of blood cleared of drug. Specifically, the elimination is quantified in terms of the equivalent volume of the blood that would be cleared completely to match the extraction observed. Take, for example, a solution that flows through an extraction unit at a rate of 1 L/h. If the concentration entering is 10 mg/L and the concentration leaving after extraction is 5 mg/L, 5 mg/L is extracted. This degree of extraction is equivalent to clearing completely 50% of the fluid entering. The clearance is 0.5 × 1 L/h = 0.5 L/h. Further, it can be seen that the rate of elimination in the example above is Cl · C or 0.5 L/h × 10 mg/L = 5 mg/h.
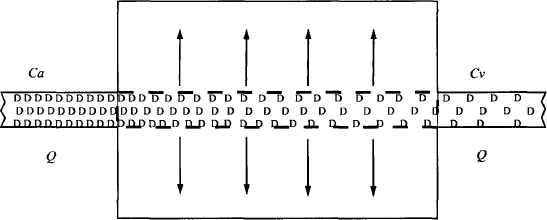
Clearance is a function of both the blood flow to the organ and the efficiency with which the processes within the liver or kidney can eliminate a drug. Its characteristics and determinants are most conveniently presented using a hypothetical extraction unit to represent an organ of elimination (Figure 5.2). A solution of drug flows through this unit at a rate of Q(L/h). The concentration of drug in the incoming solution is Ca(mg/L) and the concentration leaving is Cv (mg/L). As a result of the elimination/extraction, Ca > Cv.
FIGURE 5.2 Extraction unit. Drug (D) in solution is extracted as it flows through the unit. The concentration of drug entering the extraction unit is Ca (mg/L) and the concentration of drug leaving the unit is Cv (mg/L). The solution flows through at a rate of Q (L/h).
The rate of presentation of the drug to the extraction unit is expressed as
(5.6) 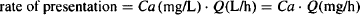
The rate of which the drug leaves the extraction unit is
(5.7) 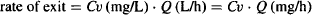
The rate of extraction or elimination is calculated as
(5.8) 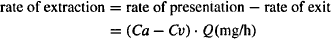
The fraction of drug entering that is extracted, the extraction ratio,
(5.9) 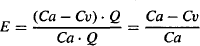
The fraction extracted, or fraction of incoming drug that is removed, E, is a measure of the unit’s ability to extract a given drug. It is independent of Ca and is a constant (elimination is first-order) that reflects the ability of the unit to extract drug that is presented to it.
If E = 1, 100% of the incoming drug is extracted or eliminated; that is, the unit possesses exceptional ability to extract this particular drug. Thus, clearance in this case is Q L/h (all the solution entering is completely cleared of drug). Mathematically, we have
(5.10) 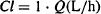
If E = 0.5, 50% of drug entering the unit is removed; that is, the unit possesses moderate ability to extract this particular drug. As the solution flows through the unit, the concentration of the drug would fall by 50%. Reducing the concentration by 50% is equivalent to removing drug completely from half of the solution as it flows through the extraction unit:
(5.11) 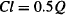
So if E = 0.3, 30% of the incoming drug is eliminated. This is equivalent to clearing 30% of the solution of drug:
(5.12) 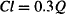
More generally,
(5.13) 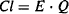
Thus, clearance is a function of the ability of the organ to eliminate a given drug (E) and the blood flow (Q) to the organ. Clearance can never exceed the blood flow to the organ. To make these principles clearer, they can be illustrated further by an example.
Example 5.1 Consider an extraction unit, where Ca = 200 mg/L, Cv = 140 mg/L, and Q = 2 L/h (Figure E5.1). Table E5.1 shows the step-by-step determination of extraction ratio, clearance, and rate of elimination for this example and for the general case.
FIGURE E5.1 Extraction unit. Drug (D) in solution is extracted as it flows through the unit. The concentration of drug entering the extraction unit is 200 mg/L and the concentration of drug leaving the unit is 140 mg/L. The fluid flows through at a rate of 2 L/h.
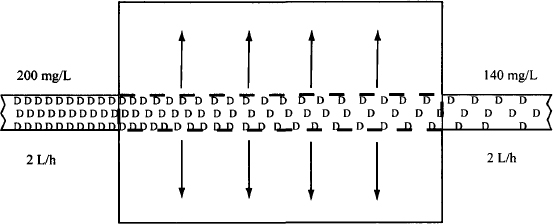
TABLE E5.1 Step-by-Step Derivation of Expressions for Clearance and the Rate of Elimination
| Extraction Unit | General Expressiona | |
| Rate of presentation of drug | 200 × 2 = 400 mg/h | Q · Ca |
| Rate drug exits | 140 × 2 = 280 mg/h | Q · Cv |
| Rate of extraction | 400 − 280 = 120 mg | (Ca − Cv) · Q |
| Fraction extracted: extraction ratio (E) | 120/400 = 0.3 or 30% | (Ca − Cv)/Ca |
| Clearance (volume of fluid cleared completely per unit time) | 2 L/h × 0.3 = 0.6 L/h | Q · E |
| Rate of extraction or elimination | 0.6 L/h × 200 mg/L = 120 mg/h | Cl · Ca |
| a Ca and Cv are the concentrations entering and leaving the extraction unit, respectively, Q is the blood flow, E is the fraction of the drug entering in the solution that is extracted, and Cl is the clearance of the extraction unit. | ||
5.2.2 Total Clearance, Renal Clearance, and Hepatic Clearance
Total clearance (Cl), which is referred to as total body clearance or systemic clearance, is sum of all the component clearances:
(5.14) 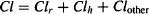
where Cl is the total body clearance, Clr the renal clearance, Clh the hepatic clearance, and Clother represents any other form of clearance. As we demonstrate later in the chapter, total body clearance and renal clearance, are easily measured, and values for specific drugs are usually widely available. In contrast, other forms of clearance, including hepatic clearance, are much more difficult to quantify. As a result, clearance is often expressed
(5.15) 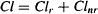
where Clnr, which is the sum of all nonrenal forms of clearance, is the difference between measured total body clearance and renal clearance.
As discussed previously, hepatic clearance is the major form of nonrenal clearance, and as a result,
(5.16) 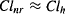
and the expression for total clearance is often presented as
(5.17) 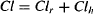
In summary:
5.2.3 Relationships Among Clearance, Volume of Distribution, Elimination Rate Constant, and Half-Life
Renal excretion and hepatic metabolism are first-order processes, and the rate of elimination can be expressed using first-order kinetics:
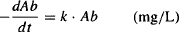
But elimination can also be expressed using clearance (Cl):
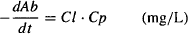
Combining equations (5.18) and (5.19) gives us
(5.20) 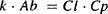
But Ab − Cp · Vd:
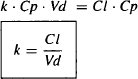
Since t1/2 = 0.693/k
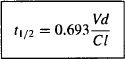
Equations (5.21) and (5.22) show the relationship between the two parameters for the rate of drug elimination and the parameters for elimination (clearance) and distribution (volume of distribution). The boxes around equations (5.21) and (5.22) indicate they are important and should be memorized.
5.2.4 Primary and Secondary Parameters
It is very important to distinguish between the dependent (secondary parameters) and independent (primary parameters) in equations (5.21) and (5.22). Clearance and volume of distribution are the primary pharmacokinetics parameters for elimination and distribution, respectively. Primary parameters are independent parameters. Thus, clearance will not change if distribution changes, and volume of distribution will not change if elimination changes. In contrast, the elimination rate constant and the half-life are secondary or derived parameters. They are dependent on the primary parameters of clearance and volume of distribution, and their value will change with changes in elimination (Cl) and/or distribution (Vd). As derived parameters, the elimination rate constant and half-life cannot change independent of clearance and volume of distribution, and cannot change either of these primary parameters.
It is also important to appreciate that clearance is not a measure of the rate of drug elimination. It is one of the two factors (Vd is the other) that determine how quickly or slowly a drug is eliminated from the body. Many drugs that have high values of clearance are eliminated rapidly (short half-lives). For example, buspirone, didanosine, metoprolol, and morphine all have high clearances and short half-lives in the region of 1 to 2 h. Similarly, many drugs that have low clearances are eliminated slowly; for example, phenobarbital has a very low clearance and a half-life of around 4 days. But a drug’s volume of distribution is also a factor. For example, diltiazem and felodipine have similar values for clearance (around 800 mL/min), but felodipine’s elimination half-life is about four times larger than diltiazem’s because its volume of distribution is about four times larger than that of dilitazem. Also, chloroquine and amiodarone both have moderate clearances but have exceptionally long half-lives (~one month) because they both have very large volumes of distribution (~ 14,000 and 4,500 L, respectively). In both cases the majority of the drug in the body is located in the tissues and is inaccessible to the organs of elimination.
5.3 RENAL CLEARANCE
In the kidney the parent drug may be eliminated by excretion into the urine. A drug’s renal clearance is a measure of the efficacy with which the kidney can accomplish this process. The value of renal clearance is determined by blood flow to the part of the kidney involved in drug excretion and on the ability of the kidney to excrete the drug. The ability of the kidney to excrete the drug is a function of renal physiology and the physicochemical properties of the drug.
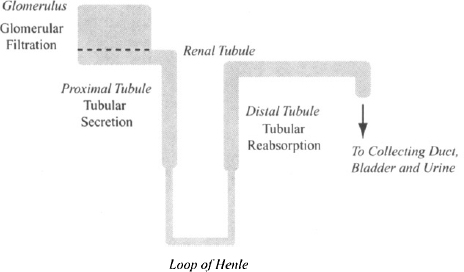
The nephron is the functioning unit of the kidney, and each kidney possesses about 1 to 1.5 million of these. A simplified diagram of the nephron is shown in Figure 5.3. In the glomerulus, plasma water is filtered (glomerular filtration) into the renal tubule, the contents of which eventually drain into the bladder. However, as the filtrate passes through the tubule, components such as water and dissolved substances, including drugs, may move back and forth across the renal tubule membrane between the blood and the lumen of the tubule. The movement of compounds from the capillaries surrounding the tubules (peritubular capillaries) into the tubule is referred to as tubular secretion The movement in the opposite direction, from the tubules back into the blood, is referred to as tubular reabsorption During transit through the tubule, much of the plasma water is reabsorbed back into the bloodstream.
FIGURE 5.3 Simplified diagrammatic representation of the nephron. Three processes in the kidney participate in drug excretion: glomerular filtration in the glomerulus; tubular secretion, which takes place primarily in the proximal renal tubule; and tubular reabsorption, which occurs primarily in the distal renal tubule.
5.3.1 Glomerular Filtration
About 20% of the renal blood supply (about 1.2 L/min) is directed to the glomerulus. In the glomerulus the blood is subjected to hydrostatic pressure, which forces plasma water and small solutes, including most drugs, through the capillary membrane and into the renal tubule. The glomerular capillaries are extremely permeable and permit the free passage of neutral molecules below 4 nm in diameter. The filtration of compounds with diameters between 4 and 8 nm is inversely proportional to their size. Compounds greater than 8 nm are excluded completely. The glomerular capillary wall appears to possess a negative charge, which repels negatively charged molecules. As a result, the permeability of anions is less than that of neutral molecules. Plasma proteins, including albumin, which has a negative charge and a diameter of about 7 nm, do not undergo any appreciable filtration (2). Small proteins such as insulin (MW ~ 5000 Da) are able to undergo filtration.
The normal glomerular filtration rate (GFR) in the standard 70-kg young adult male is about 120 to 130 mL/min per 1.73 m2 (about 7.5 L/h or 180 L/day), and it declines with age. If a drug does not bind to plasma proteins and it is small enough to be filtered in the glomerulus, its clearance by glomerular filtration is equal to the glomerular filtration rate (at this point, the filtrate, and any drug it contains, is eliminated and removed from the circulation). It represents the volume of plasma completely cleared of drug:
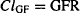
where CLGF is clearance by glomerular filtration.
However, many drugs bind to the plasma proteins, and bound drug will not be filtered. For example, if a drug is bound completely (100%), its clearance by glomerular filtration will be zero. If, on the other hand, a drug is 70% bound to the plasma proteins (fu = 0.3), only 30% of the drug in the plasma will be filtered and
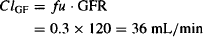
In summary:
(5.23) 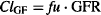
5.3.2 Tubular Secretion
Drug elimination can be augmented further by tubular secretion, a process that results in the movement of drug from the blood in the peritubular capillaries that surround the tubule into the lumen of the tubule. In many cases, this process is accomplished by the action of transporters, which appear to be concentrated in the proximal tubular cells. Several reviews of the role of transporters in renal excretion have been written (3–5). A summary of this material is presented below.
Uptake transporters (OAT1, OAT3, and OCT2) are present on the basolateral side of the renal tubular membrane, and efflux transporters (P-gp, MRP2, and MRP4) are present on the apical side (Figure 5.4). The two transporter systems can work together to facilitate tubular secretion and the excretion of drugs. The uptake transporters initiate the process and move the drug from the blood into the renal tubular membrane. The efflux transporters can then conclude tubular secretion by carrying the drug into the tubule. The tubular secretion of both fexofenadine, which is a substrate for renal OAT3 and P-gp (6, 7), and that of adefovir, which is a substrate for OAT1 and MRP4, appears to be brought about in this manner. Substrates of the renal OAT transporters include β-lactam antibiotics, diuretics (furosemide, bendroflumethiazide), nonsteroidal antiinflammatory drugs (e.g., aspirin, indomethacin), antiviral drugs (e.g., acyclovir, cidofovir, zidovudine), and methotrexate. There is some overlap in the substrate specificity of the OATs, but OAT1 appears to be more important for small molecular mass drugs such as adefovir, and OAT3 appears to be more important for larger drugs such as the penicillin G and for sulfate and glucoronide conjugates such as estradiol glucoronide. OAT3 can also carry positively charged molecules, such as the H2-receptor antagonists (famotidine), and molecules with both positive and negative charges, such as fexofenadine. The high drug concentrations in the renal tubular cells produced by uptake transporters have been implicated in promoting the renal toxicity of drugs, such as adefovir, cidofovir, and cephaloridine. This raises the possibility that the renal toxicity of these drugs could be reduced by the coadministration of inhibitors of the OAT transporters, such as probenecid.
FIGURE 5.4 Drug transporters in the human renal tubule. The uptake transporters (U) organic cation transporter (OCT2) and organic anion transporters (OAT1 and OAT3) are found on the basolateral side of the renal tubular membrane. These transporters promote excretion by transporting drugs from the blood into the tubular cells. The efflux transporters (E), permeability glycoprotein (P-gp) and multidrug resistance–associated protein family (MRP2 and MRP4) can conclude the process by transporting drugs from the tubular cells into the tubule.
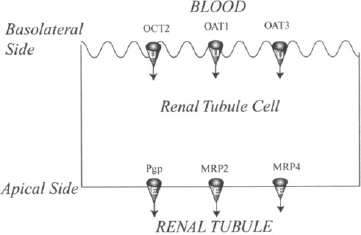
Substrates for OCT2 include the H2 antagonists (cimetidine, famotidine), cisplatin, and metformin, which is almost completely eliminated by renal excretion. OCT2-mediated uptake of cisplatin is also believed to be an important factor in its renal toxicity. In support of this, cimetidine, an OCT2 inhibitor, was found to reduce the severity of the toxicity.
Modification of activity of the uptake transporters by concomitant medications is an important source of drug–drug interactions for drugs that are primarily cleared renally. Probenecid, which inhibits OAT, reduces the clearance of many drugs, including the penicillins, the quinolone antibiotics, and fexofenadine (7). This interaction has been used to advantage to prolong the half-life of penicllins. In the case of methotrexate, inhibition of OAT by either probenecid or penicillin is associated with increased methotrexate-induced bone marrow suppression. The coadministration of metformin with cimetidine, which is both a substrate and an inhibitor of OCT2, decreases the renal clearance of metformin.
The role of efflux transporters in drug excretion is less clear at this time. P-gp, MRP2, and MRP4 are all present in the renal tubular membrane, and it is probable that they contribute to the excretion of their substrate drugs. Digoxin is often used as a model substrate for P-gp because it does not undergo any metabolism by CYP3A4. As discussed in Chapter 3, digoxin is also a substrate for intestinal P-gp. In theory, inhibition of P-gp could increase the absorption of digoxin in the intestine and/or decrease its elimination in the kidney. Both would increase the serum concentrations of digoxin. Rifampin, which induces P-gp, was found to have a greater effect on the intestinal efflux of digoxin than on the renal efflux. In other studies, the relative contribution of altered renal and/or intestinal P-gp on digoxin’s pharmacokinetics is not clear. From a clinical standpoint, modifiers of P-gp should be used cautiously with digoxin. Irrespective of the mechanism, inhibitors and inducers may increase and decrease, respectively, the body’s exposure to digoxin. MRP2 in the renal tubule appears to be involved in the elimination of methroxeate, and studies in mice suggest that MRP4 is involved in the excretion of hydrocholorothiazide, furosemide, some cephaloridins (ceftizoxime and cefazolin), adefovir, and tenofovir.
5.3.3 Tubular Reabsorption
As the filtrate moves through the proximal tubule and the loop of Henle, water is reabsorbed back into the systemic circulation. When the filtrate reaches the distal tubule, about 80% of the filtered water has been reabsorbed. As a result, the concentration of drugs in the filtrate becomes higher than that in the blood in the surrounding capillaries, and drugs diffuse along their concentration gradient back into the systemic circulation. The extent of this tubular reabsorption is controlled by:
(5.24) 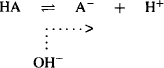
(5.25) 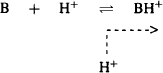
5.3.4 Putting Meaning into the Value of Renal Clearance
A drug’s overall renal clearance is a function of glomerular filtration, tubular secretion, and tubular reabsorption. It may be expressed as

where ClTS is clearance by tubular secretion and TR represents tubular reabsorption.
Example 5.2 Consider a hypothetical drug that does not bind to the plasma proteins (fu = 1) and has a renal clearance of 50 mL/min (Figure E5.2). Use this information to gain insight into the processes involved in its renal clearance.



