Phase I reactions add or unmask functional groups (e.g., –OH, −NH2, −SH, −COOH, etc.), which can then participate in phase II reactions. These are non-synthetic reactions. Oxidation reactions occur mainly on the smooth endoplasmic reticulum (sER) and are catalyzed by a family of enzymes known as microsomal “mixed-function oxidases” (MFO, so called because they catalyze a large range of oxidation reactions on a variety of substrates). These microsomal oxidative enzymes are also known as “monooxygenases”. Other phase I reactions (including some oxidation reactions) take place in the cytosol, plasma and mitochondria. Phase I metabolites are usually not much more polar than their parent drugs, but they are often chemically reactive and may even be toxic (e.g., acetaminophen-induced liver toxicity or cyclophosphamide-induced bladder toxicity).
Phase II reactions are synthetic and involves the addition of a large endogenous substrate to the drug or its phase I metabolite to form a conjugate. Thus, phase II metabolites are frequently much more polar and almost always inactive, except for a few drug metabolites (e.g., morphine 6-glucuronide, N-acetylprocainamide or minoxidil sulfate). These conjugation reactions occur mainly in the cytosol, except for glucuronide conjugation reactions, which occur on sER and thus glucuronyl-transferases are microsomal enzymes. Generally speaking, only the microsomal enzymes are inducible.
Drugs may be excreted without being metabolized (e.g., gentamicin). However, the majority of drugs undergo some form of metabolism (whether phase I or phase II, or more commonly, both) before excretion. While most drugs undergo phase I and phase II reactions in that order, in some cases (e.g., isoniazid), phase II reactions precede phase I reactions (Fig. 5.1).
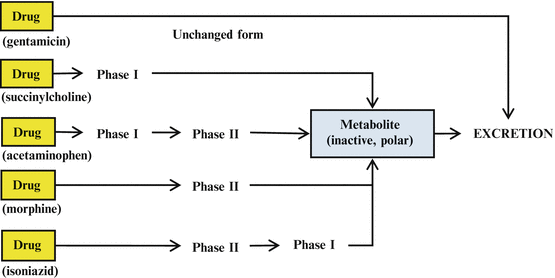
Fig. 5.1
Schematic diagram showing different pathways of drug metabolism
Cytochrome P450
Cytochrome P-450 (P450) is a heme-containing enzyme, which is part of the MFO system, and can exist in many isoforms (e.g., CYP1A2, CYP2D6, CYP3A4). These P450 enzymes are predominantly expressed in the liver and are extremely important in drug metabolism. They are essential for most drug metabolism, contribute to most of the observed variations in drug response of differing ethnic origins (due to genetic polymorphism of P450 isozymes), account for many of the important drug interactions (Table 5.2), and are responsible for a number of serious adverse effects. In view of this, physicians should be cautious in prescribing drugs known to be P450 inducers or inhibitors, especially to critically ill patients.

Table 5.2
Important P450 enzymes and examples of drugs which require dose adjustment to prevent adverse effects resulting from drug interaction involving enzyme inhibition or induction
Factors Affecting Drug Metabolism
Both internal (e.g., age, diseases or genetic factors) and external (e.g., diet or environment) factors may affect the rate or extent of drug metabolism.
1.
Age: The activity of hepatic microsomal enzymes is low in both the very young and the elderly patients resulting in impaired drug metabolism. Hence there is an increased risk of drug toxicity with reduced inactivation of drugs. For example, the grey baby syndrome associated with the use of chloramphenicol in neonates is the result of an inadequate amount of glucuronyl transferases to conjugate the drug in the immature liver. On the other hand, phenobarbital, a potent enzyme inducer, can be given to accelerate the clearance of bilirubin in jaundiced neonates by enhancing bilirubin conjugation.
2.
Diseases: Chronic liver diseases (e.g., cirrhosis) markedly affect the hepatic metabolism of certain drugs (e.g., diazepam) and prolong their duration of action in the body, leading to potential risk of overdose. Therefore, drugs which are highly dependent on liver for their elimination (e.g., opioids, acetaminophen, and propranolol) should be used with caution in patients with liver diseases and their dosages reduced accordingly.
3.
Genetic variation: The metabolism of certain drugs (e.g., succinylcholine and isoniazid) is significantly reduced in susceptible people who have either a defective enzyme (e.g., atypical pseudocholinesterase for hydrolyzing succinylcholine) or abnormally low level of an enzyme (e.g., acetylation of isoniazid).
4.
Diet: Certain plant foods (e.g., cabbage and cauliflower) and barbecued foods contain bioactive compounds which are enzyme inducers. Regular ingestion of such foods can significantly increase the metabolism of administered drugs and reduce their therapeutic effectiveness. On the other hand, grapefruit juice is known to inhibit certain P450 enzymes that metabolize a number of drugs and may result in increased risk of toxicity.
5.
Environment: Cigarette smoke and dichlorodiphenyltrichloroethane (DDT, a banned insecticide in many countries) are also powerful inducers of drug-metabolizing enzymes.
Drug Elimination by Excretion
The metabolism of drugs usually results in water soluble metabolites which are readily excreted. Some drugs, however, are directly eliminated without metabolism. While the liver is the major organ for eliminating drugs by metabolism, it is the kidneys which are responsible for eliminating most drugs and metabolites by excretion. Besides the kidneys, the intestine, the biliary system and the lungs are also involved in excreting many drugs, while a small amount of drugs may be excreted in the saliva, sweat and milk.
Renal Excretion
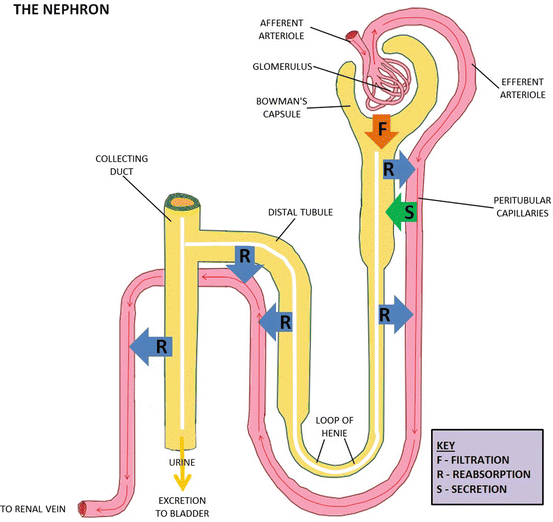
Three processes are involved in the renal excretion of drugs (Fig. 5.2). They are glomerular filtration, active tubular secretion and passive tubular reabsorption. While the former two processes facilitate drug excretion and are not affected by urine pH, the latter one decreases drug excretion and is pH-dependent.