Fig. 4.1
A schematic diagram showing the flow of solutes from blood to surrounding fluids and tissue-cells. Drug A, being more lipid soluble than Drug B, enters both interstitial (ISF) and intracellular fluids (ICF), whereas the polar Drug B, is only able to cross over into the interstitial fluid
How Do Drugs Traverse Blood-Tissue Barriers
The ease with which drugs cross blood-tissue barrier depends on the structural and functional characteristics of the capillary endothelial cells at those tissue sites (Fig. 4.2). In most of the capillary beds, such as those of muscle, fat and nervous tissues, the capillary endothelium lacks pores and the cells are joined by tight non-permeable junctions. In other capillary beds, such as those of heart muscle, the endothelial cell allows the transport of fluid or macromolecules (e.g., insulin) in vesicles from the blood into the interstitium and vice versa. In some capillary beds, such as in the pancreas, gut, kidney glomeruli and endocrine glands, the endothelial lining behaves as though it were fenestrated by pores. In the liver, spleen and bone marrow, open spaces exist between endothelial cells, called intercellular clefts, which allow free exchange of drugs (most of which have molecular weights of less than 900) between blood and the interstitium.
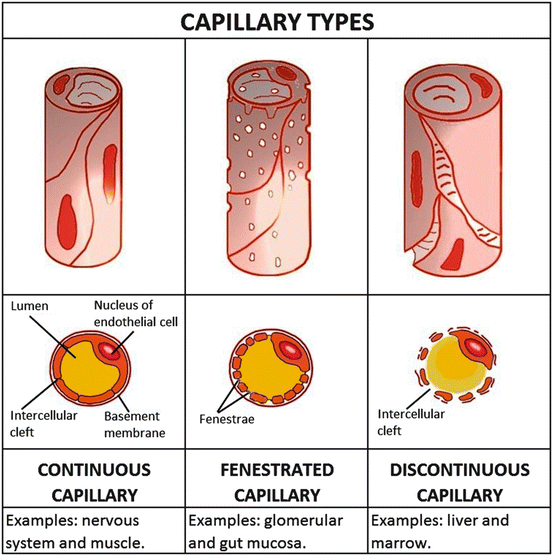
Fig. 4.2
Different types of capillaries with varying degrees of permeability, their respective cross-sections and examples of typical locations where these capillary types are found
In summary, the capillary wall basically consists of an endothelial cell layer and a basement membrane enveloping it. The permeation of drugs across this capillary wall can occur by one of several processes such as passive diffusion, transcytosis (vesicular transport) and bulk flow as well as by carrier-mediated transport mechanisms such as facilitated diffusion or active transport.
Factors Affecting the Rate of Drug Distribution
After a drug enters the bloodstream, its rate of distribution to the various body tissues is governed predominantly by two factors, each of which is potentially rate-limiting: (1) the rate of blood flow (perfusion) to the tissue and (2) the ability of the drug to cross membranes (permeability) to enter the tissue.
For drugs which have high membrane permeability (e.g., thiopental, steroids and benzodiazepines), the rate of distribution to tissues depends mainly on the rate of blood flow to these tissues. Thus, tissue drug concentration would increase most rapidly in highly perfused tissues such as kidneys, liver, lung, heart and brain, more slowly in less well perfused tissues such as skeletal muscle, but very slowly in poorly perfused tissues, such as bone, fat and skin. The distribution of these lipophilic drugs is described as perfusion-limited.
On the other hand, drugs with poor membrane permeability (e.g., penicillins, aminoglycoside antibiotics, succinylcholine and vecuronium) would distribute more readily to tissues such as bone marrows, spleen, liver and muscle, where the capillary wall is more “leaky”, than to the brain, where the capillary wall seems to be impermeable. This selective distribution occurs despite a large amount of the drug being delivered to the highly perfused brain, and is an example of permeability-limited drug distribution.

Factors Affecting the Extent of Drug Distribution
Distribution is a reversible process. Hence, the exchange of drug molecules across a barrier would continue until the concentrations of the free drugs on both sides of the barrier are the same. While tissue perfusion has a major effect on the rate of distribution of drugs to various tissues, drug properties and membrane permeability play a greater role on the extent of drug distribution at equilibrium.
Important physicochemical properties of drugs which affect their extent of distribution include molecular size, degree of ionization, octanol:water partition coefficient and their relative affinities for tissue and plasma proteins.
Drugs with large molecular weight (e.g., heparin) and those which are extensively bound to plasma protein (e.g., warfarin) have difficulty crossing the capillary wall into the interstitium. Their distribution would be restricted to mainly the plasma compartment (3 L/70 kg). Polar drugs such as gentamicin and vecuronium cross the capillary endothelium readily (by bulk flow) into the extravascular fluid compartment (14 L/70 kg) but cannot diffuse into the tissue cell. In contrast, lipophilic drugs such as diazepam and theophylline diffuse easily into intracellular fluid compartment and distribute into total body water (42 L/70 kg). Some drugs (e.g., digoxin and chloroquine) bind extensively to extravascular tissue proteins resulting in volume of distribution (Vd) values that far exceed the total body water volume. This phenomenon can be observed even with drugs (e.g., fluoxetine and imipramine) which have extensive plasma protein binding (Table 4.1).
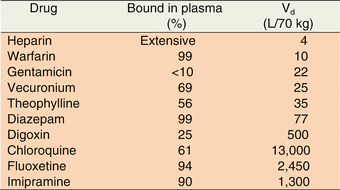
Table 4.1
Volumes of distribution (Vd) and degree of plasma protein binding for selected drugs
The volume of distribution (Vd) is the second most important pharmacokinetic parameter after plasma clearance (CL). It determines the loading dose (DL) to be given during multiple dosing in order to achieve plasma steady-state concentration (Css) faster, according to the equation DL = VdCss. An obvious consequence of having a large Vd is that a large loading dose is needed for this purpose.
Ageing is accompanied by changes in body fat, lean body mass and total body water. These changes result in reduced Vd of water soluble drugs, e.g., digoxin, (which may lead to increased initial drug concentration) and increased Vd of lipophilic drugs, e.g., benzodiazepines (which may lead to increased elimination half-life and prolonged effect). Both types of drug may therefore require a reduction in dose and/or dose interval.
Binding of Drugs to Plasma Proteins
In the plasma, drugs may bind to various proteins, primarily albumin but also to α1-acid glycoprotein, globulins (low capacity but high affinity) and lipoproteins. This binding is reversible and occurs to varying extents for different drugs (e.g. <10 % for gentamicin to ≥99 % for warfarin and diazepam). Many acidic drugs, such as salicylates and penicillins, and some neutral or basic drugs bind to albumin (low affinity but high capacity), while α1-acid glycoprotein binds mainly basic drugs such as lignocaine and propranolol. Plasma globulins (α, β, γ-globulins) have low capacity but high affinity for the binding of endogenous substances such as corticosteroids.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


