The administration of controlled amounts of the purified active compound removed biological variability in the potency of the crude plant preparation.
 The administration of the active component removed the unwanted and potentially toxic effects of contaminating substances in the crude preparations.
The administration of the active component removed the unwanted and potentially toxic effects of contaminating substances in the crude preparations. The identification and isolation of the active component allowed the mechanism of action to be defined, leading to the synthesis and development of chemically related compounds based on the structure of the active component but with greater potency, higher selectivity, fewer unwanted effects, altered duration of action and better bioavailability. For example, chemical modification of salicylic acid by acetylation produced acetylsalicylic acid, or aspirin, first marketed in 1899, with greater analgesic and antipyretic activity and lower toxicity than the parent compound.
The identification and isolation of the active component allowed the mechanism of action to be defined, leading to the synthesis and development of chemically related compounds based on the structure of the active component but with greater potency, higher selectivity, fewer unwanted effects, altered duration of action and better bioavailability. For example, chemical modification of salicylic acid by acetylation produced acetylsalicylic acid, or aspirin, first marketed in 1899, with greater analgesic and antipyretic activity and lower toxicity than the parent compound.Thus, although drug therapy has natural and humble origins, it is the application of scientific principles, particularly the use of controlled experiments and clinical trials to generate reliable knowledge of drug actions, which has given rise to the clinical safety and efficacy of modern medicines. In the age of ‘scientific reason’ it is surprising that so many people believe that ‘natural’ medicinal products offer equivalent therapeutic effectiveness with fewer unwanted effects.
A major advantage of modern drugs is their ability to act selectively; that is, to affect only certain body systems or processes. For example, a drug that both lowers blood glucose and reduces blood pressure may not be suitable for the treatment of someone with diabetes mellitus (because of unwanted hypotensive effects) or a person with hypertension (because of unwanted hypoglycaemic effects), or even of those with both conditions (because different doses may be needed for each effect).
Drug discovery
The discovery of a new drug can be achieved in several different ways (Fig. 3.1). The simplest method is to subject new chemical entities (novel chemicals not previously synthesised) to a battery of screening tests that are designed to detect different types of biological activity. These include in vitro studies on isolated tissues, as well as in vivo studies of complex and integrated systems, such as animal behaviour. Novel chemicals for screening may be produced by direct chemical synthesis or isolated from biological sources, such as plants, and then purified and characterised. This approach has been revolutionised in recent years by developments in high-throughput screening (or HTS), which takes advantage of laboratory robotics for liquid handling combined with in vitro cell lines expressing cloned target proteins in tiny reaction volumes in microplates containing hundreds or thousands of reaction wells. Active compounds, which may be small-molecule libraries derived from bacterial or fungal sources, or proteins derived from solid-phase peptide synthesis, can then be selected based on interactions with cells that express a range of possible sites of action, such as G-protein-coupled or nuclear receptors or enzymes important in drug metabolism. Such methods allow the screening of many hundreds of compounds each day and the selection of suitable ‘lead compounds’, which are then subjected to more labour-intensive and detailed tests.
A second approach involves the synthesis and testing of chemical analogues and modifications of existing medicines; generally, the products of this approach show incremental advances in potency, selectivity and bioavailability (structure–activity relationships). However, additional or even new properties may become evident when the compound is tried in animals or humans; for example, minor modifications of the sulphanilamide antimicrobial molecule gave rise to the thiazide diuretics and the sulfonylurea hypoglycaemics.
More recently, attempts have been made to design substances to fulfil a particular biological role, which may entail the synthesis of a naturally occurring substance (or a structural analogue), its precursor or an antagonist. Good examples include levodopa, used in the treatment of Parkinson’s disease, the histamine H2 receptor antagonists and omeprazole, the first proton pump inhibitor. Logical drug development of this type depends on a detailed understanding of human physiology both in health and disease. High-throughput screening is particularly useful in such a focused approach. In silico (computer-based) approaches to the modelling of receptor binding sites have facilitated the development of ligands with high binding affinities and, often, high selectivity.
The recent phenomenal advances in molecular biology have led to the increasing use of genomic techniques, both to identify genes associated with pathological conditions and subsequently to develop compounds that can either mimic or interfere with the activity of the gene product. Such compounds are often proteins, which gives rise to problems of drug delivery to the relevant tissue and to the site of action, which may be intracellular, and also raises issues related to safety testing (see below). A good example of the potential of genomic research is the drug imatinib (Ch. 52), which was developed to inhibit the Bcr-Abl receptor tyrosine kinase, which was implicated in chronic myeloid leukaemia cells by molecular biological methods; imatinib is a non-protein organic molecule with a high oral bioavailability.
Irrespective of the approach, drug development is a long and costly process, with estimates of approximately 14 years and over GB£500 million to bring one new drug to market. Much of this cost lies in gaining the preclinical and clinical evidence required for approval of a new drug by regulatory bodies.
Drug approval
Each year, many thousands of new chemical entities and also compounds purified from plant and microbial sources are screened for useful and novel pharmacological activities. Potentially valuable compounds are then subjected to a sequence of in vitro and in vivo animal studies and clinical trials in humans, which provide essential information on safety and therapeutic benefit (Fig. 3.2).
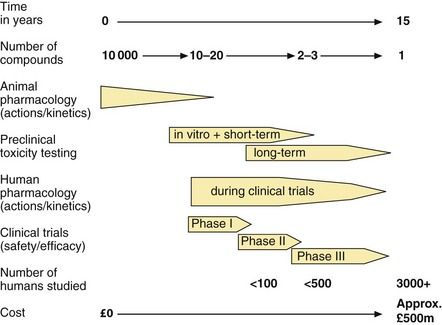
Fig. 3.2 The development of a new drug to the point at which a licence is approved.
Post-marketing surveillance will continue to add data on safety and efficacy.
All drugs and formulations licensed for sale in the UK have to pass a rigorous evaluation of:



In the European Union (EU), new drugs are approved under a harmonised procedure of drug regulation. The European Medicines Agency (EMA; www.ema.europa.eu/ema) is a decentralised body of the EU with headquarters in London and is responsible for the regulation of medicines within the EU. It is broadly comparable to the Food and Drug Adminstration (FDA; www.fda.gov) in the USA. The EMA receives advice from the Committee for Medicinal Products for Human Use (CHMP), which is a body of international experts who evaluate data on the safety, quality and efficacy of medicines. Other EMA committees are involved in evaluating paediatric medicines, herbal medicines and advanced therapies such as gene therapy. Under the current EU system, new drugs are evaluated by the CHMP and national advisory bodies have an opportunity to assess the data before a final CHMP conclusion is reached.
The UK Commission on Human Medicines (CHM) was established in 2005 to replace both the Medicines Commission (MC) and the Committee on Safety of Medicines (CSM), which previously had evaluated medicines regulated in the UK under the Medicines Act (1968). The CHM is one of a number of committees established under the Medicines and Healthcare products Regulatory Agency (MHRA; www.mhra.gov.uk). The MHRA provides advice to the Secretary of State for Health.
Safety
Historically, the introduction of new drugs has been bought at a price of significant toxicity, and regulatory systems have arisen as much to protect patients from drug toxicity as to ensure benefit. In the USA, the FDA was established in 1937, following a dramatic incident in which 76 people died of renal failure after taking an elixir of sulphanilamide which contained the solvent diethylene glycol. Similarly, some 30 years later, the occurrence of limb malformations (phocomelia) and cardiac defects in infants born to mothers who had taken thalidomide for the treatment of nausea in the first trimester of pregnancy led to the establishment of the precursor of the CSM in the UK.
Today, major tragedies are avoided by a combination of in vitro studies and animal toxicity tests (preclinical testing) and by careful observation during clinical studies on new drugs (see below). The development and continuing refinement of preclinical toxicity testing has increased the likelihood of identifying chemicals with direct organ toxicity. During clinical trials, immunologically mediated effects are likely to be seen at the lower end of the dose ranges that are used in such trials (see Ch. 53).
Quality
An important function of regulatory bodies is to ensure the consistency of prescribed medicines and their manufacturing processes. Drugs have to comply with defined criteria for purity and limits are set on the content of any potentially toxic impurities. The stability and, if necessary, sterility of the drug also have to be established. Similarly, licensed formulations must contain a defined and approved amount of the active drug, released at a specified rate. There have been a number of cases in the past in which a simple change to the manufactured formulation affected tablet disintegration, the release of drug and the therapeutic response. The quality of drugs for human use is defined by the specifications in the European Pharmacopoeia (Ph.Eur.) and the British Pharmacopoeia (BP).
Efficacy
All medicines, apart from homeopathic products, must have evidence of efficacy for their licensed indications. Efficacy, i.e. the ability to produce a predefined level of clinical response, can be established only by trials in people with the disease, for whom the medicine is intended, and therefore the demonstration of efficacy is a major aim of the later phases of clinical research (Fig. 3.2).
Establishing safety and efficacy
Regulatory bodies such as the CHMP and CHM require supporting data from in vitro studies, animal studies and clinical investigations before a new drug is approved. Although there is some overlap, the basic aims and goals are:
 preclinical studies: to establish the basic pharmacology, pharmacokinetics and toxicological profile of the drug and its metabolites, using animals and in vitro systems,
preclinical studies: to establish the basic pharmacology, pharmacokinetics and toxicological profile of the drug and its metabolites, using animals and in vitro systems, phase I clinical studies: to establish the human pharmacology and pharmacokinetics, together with a simple safety profile,
phase I clinical studies: to establish the human pharmacology and pharmacokinetics, together with a simple safety profile, phase II clinical studies: to establish the dose–response relationship and to develop the dosage protocol for clinical use, together with more extensive safety data,
phase II clinical studies: to establish the dose–response relationship and to develop the dosage protocol for clinical use, together with more extensive safety data, phase III clinical studies: to establish the efficacy and safety profile of the drug in people with the proposed disease for which the drug will be indicated,
phase III clinical studies: to establish the efficacy and safety profile of the drug in people with the proposed disease for which the drug will be indicated,
Preclinical studies
Preclinical studies must be carried out before a compound can be administered to humans. These studies investigate three areas:
 pharmacological effects: in vitro effects using isolated cells, tissues or organs; receptor-binding characteristics; in vivo effects in animals and/or animal models of human diseases; prediction of potential therapeutic use,
pharmacological effects: in vitro effects using isolated cells, tissues or organs; receptor-binding characteristics; in vivo effects in animals and/or animal models of human diseases; prediction of potential therapeutic use, pharmacokinetics: identification of metabolites (since these may be the active form of the compound); evidence of bioavailability (to assist with the design of both clinical trials and in vivo animal toxicity studies); establishment of principal route and rate of elimination,
pharmacokinetics: identification of metabolites (since these may be the active form of the compound); evidence of bioavailability (to assist with the design of both clinical trials and in vivo animal toxicity studies); establishment of principal route and rate of elimination,Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


