Drugs that are already either in the disperse state (suspension formulations) or in solution (solution formulations, e.g., syrups) will be farther along the absorption chain than solid dosage forms such as tablets. As a result, drugs formulated as suspensions and solutions, in particular, often display more rapid and/or more complete absorption than do their solid dosage formulations. The absorption of itraconazole, for example, which has poor dissolution properties, is better from a solution formulation than from a tablet.
3.4 EXTENT OF DRUG ABSORPTION
3.4.1 Bioavailability Factor
The extent of drug absorption is assessed by means of its bioavailability factor, which is usually referred to simply as “bioavailability,” although strictly speaking, a drug’s bioavailability refers to both the rate and extent of drug absorption. The bioavailability factor, F is the fraction of the dose that reaches the systemic circulation intact. As a factor it can achieve a value between zero (none of the dose reaches the systemic circulation) and 1 (the entire dose reaches the systemic circulation). Thus, if a drug’s F value is 0.80, 80% of the dose administered reaches the circulation as intact drug. Since the body is exposed only to the dose that is absorbed, the effective dose of a drug may be defined as the product of the bioavailability and the dose administered:
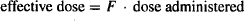
Thus, if 100 mg of a drug is administered and if F = 0.8,
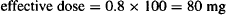
As pharmacokinetic equations are introduced throughout the book, all formulas will include the bioavailability factor as a potential qualifier of the dose. The factor will even be included in a formula specifically derived for intravenous administration, where bioavailability is always 1. This is because an intravenous formula may be applied to other routes of administration that may not have complete bioavailability, and the presence of the factor in the formula serves as a reminder to account for bioavailability if necessary.
The determinants of the bioavailability factor can be classified into three broad areas:
Under normal circumstances the bioavailability factor for a given brand is a constant and, very important, does not change with dose. A. dose-dependent bioavailability factor is an example of nonlinear pharmacokinetics, discussed in Chapter 15.
3.4.2 Individual Bioavailability Factors
A portion of the drug dose may be lost at several points during the absorption process. Overall, the losses are usually classified into three categories, which results in three types of bioavailability:
Overall bioavailability (F) may thus be expressed
(3.1) 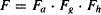
Example 3.1 Consider a drug that has the following characteristics: 20% of a dose of a drug is destroyed by acid in the stomach, the remaining drug is able to penetrate the apical membrane completely, 10% of the drug passing though the membrane is metabolized, and 40% of the drug entering the liver is metabolized. Find the effective dose when 100 mg is administered.
Solution
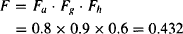
If 100 mg was administered, the effective dose would be F · D = 43.2 mg.
This categorization is useful because it separates the physicochemical factors that control Fa from the biological factors that control Fg and Fh. It is useful to distinguish Fg from Fh because they are influenced by different factors. For example, Fh may be altered by changes in hepatic blood flow, which would have no effect on Fg.
3.5 DETERMINANTS OF THE BIOAVAILABILITY FACTOR
3.5.1 Disintegration
The disintegration of the tablet into small particles suitable for drug dissolution is an essential step in the absorption process. If disintegration does not occur, or if it is very slow, the absorption of the drug will be compromised. Most tablets contain special ingredients or excipients called disintigrants (e.g., starch), which swell when wet by the gastric fluid. As a result, tablet disintegration is usually a straightforward, unproblematic step in the absorption process.
3.5.2 Dissolution
The dissolution characteristics are determined primarily by a drug’s aqueous solubility, which in turn is determined primarily by its hydrophilicity or lipophilicity, with more hydrophilic drugs dissolving in the aqueous gastrointestinal fluid more readily than do lipophilic drugs. Other factors that determine dissolution include the crystalline form of the drug and the particle size of the powdered drug. For example, the dissolution properties of poorly soluble drugs can be improved by using a process known as micronization to greatly reduce the particle size. This method is used to increase the dissolution of progesterone. Most drugs are either weak acids or weak bases and are often formulated as salts, to improve their dissolution properties. As a result of improved dissolution, the rate and extent of absorption of drugs can be increased. The specific salt form can be selected to optimize these qualities. Examples of salts include propranolol hydrochloride, phenytoin sodium, naproxen sodium, and aminophylline, which is the ethylenediamine salt of theophylline.
3.5.3 Formulation Excipients
Dosage forms contain many other ingredients in addition to active drug. These ingredients, or excipients, are pharmacologically inert and are added to assist in the manufacture of the dosage form or to impart certain properties to the finished product. For example, many powder-based dosage forms (tablets and capsules) contain lubricants to improve the flow properties of the powers so that they can be poured and transferred more easily during manufacture. Most solid dosage forms also contain diluents to increase the size of the unit dosage form, to make it a more manageable size. The importance of assessing the potential influence of inert excipients on bioavailability was demonstrated very poignantly in 1968 when over 50 Australian patients developed serious phenytoin toxicity after the manufacturer simply changed the diluent in the formulation from calcium sulfate to the more hydrophilic lactose. Other excipients that may be added to improve the properties of the final dosage form include wetting agents, dissolution enhancers, and substances to promote membrane permeation.
3.5.4 Adverse Events Within the Gastrointestinal Lumen
The gastrointestinal lumen is an extremely harsh environment. It contains enzymes, foods in various stages of digestion, and displays fairly wide changes in pH. This hostile environment can be beneficial in that it can destroy potentially harmful bacteria but can also be very detrimental for drugs.
pH The pH along the gastrointestinal tract varies from around 1.5 to 7. The gastric pH can approach about 1.5 in the presence of food as a result of gastric acid secretion stimulated by gastrin and histamine. In the fasting state, the pH generally rests at around a pH of 2 to 6. The high acidity of the stomach can be particularly problematic for drugs. The early or natural penicillins are very susceptible to acid hydrolysis, particularly penicillin G, which as a consequence is best administered parenterally. Oral preparations of penicillin V should be administered on an empty stomach (1 to 2 h before food) to minimize hydrolysis. The newer semisynthetic penicillins such as amoxicillin are much less susceptible to hydrolysis and provide better and more consistent oral bioavailability. Drugs susceptible to acid hydrolysis in the stomach can be formulated into enteric-coated tablets. The coating protects the drug from the acid in the stomach, since it will dissolve only in the higher pH of the small intestine, which generally lies in the region of 6 to 7. In another approach to protecting acid-labile drugs, antacids can be included in their formulation. This is used for the acid-labile reverse transcriptase inhibitor didanosine. The increase in the gastric pH brought about by treatment with proton pump inhibitors, H2 antagonists, and antacids can affect the dissolution and bioavailability of some drugs. The absorption of drugs such as delavirdine and the poorly soluble antifungal agents itraconazole and ketoconazole, which display much better dissolution in acidic pH, has been found to be reduced by low gastric acidity. The absorption of other drugs, such as alendronate has been found to be increased (3). The less acidic environment created by proton pump inhibitors may also reduce the absorption of calcium, which can make patients who take these drugs for extended periods more susceptible to bone fractures and osteoporosis (4).
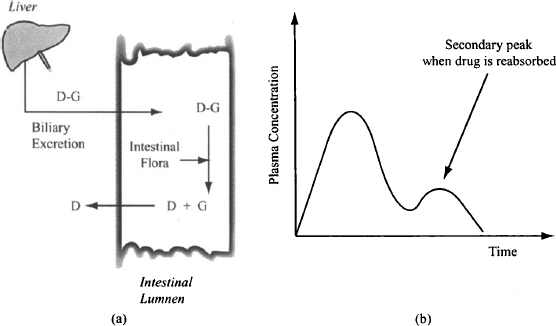
Enzymatic Attack Digestive enzymes such as pepsin, trypsin, and chymotrypsin in the gastrointestinal fluid are the main enzymes that affect drug absorption. These enzymes prevent protein drugs such as insulin from being given orally. Drugs can also be affected by the enzymes of the microflora, which are found primarily in the large intestine. Since most drugs are absorbed before they reach the large intestine, these enzymes generally do not affect the fraction of the dose absorbed initially. They can be important for drugs that are conjugated with glucuronide and subsequently excreted in the bile and then into the intestine. The gut flora can hydrolyze the conjugates and release the drug, which can then be reabsorbed back into the body. This process, known as enterohepatic recirculation (Figure 3.2), often results in the appearance of a secondary peak in the plasma concentration–time profile of a drug (Figure 3.2). The process results in the reabsorption of drug that has been eliminated from the body. Thus, in theory, enteroheaptic recirculation could result in a drug having a bioavailability factor greater than 1. The concurrent administration of broad-spectrum antibiotics such as the quinolones can reduce the population of the gut flora, which in turn can lead to reduced enterohepatic recirculation and a reduction in the body’s exposure to a drug. The enterohepatic recirculation of ethinyl estradiol, a component in oral contraceptives, can be reduced by concurrent antibiotics, which could compromise the effectiveness of the drug. Generally, this is thought to be a problem only in patients who are already predisposed to low bioavailability of ethinyl estradiol, or when the antibiotic rifampin is used. In addition to reducing the gut flora, this compound induces liver enzymes and can increase the metabolism of estradiol to further reduce the body’s exposure to estradiol.
FIGURE 3.2 Diagrammatic representation of enterohepatic recirculation (a). The glucuronide conjugate of the drug is hydrolyzed by the gut flora. The reabsorption of the drug results in a second peak on the plasma concentration–time curve (b).
Interaction with Other Components of the Gastrointestinal Fluid Drugs may interact adversely with other components of the gastric fluid, such as digested and undigested foods and other drugs. The classical example of this type of interaction is the complexation reaction between tetracycline and di- and trivalent ions (Ca2+, Mg2+, Al3+) found in dairy products and antacids. The tetracycline complex is insoluble and the drug cannot be absorbed. Similar complexes are formed with the quinolone antibiotics. Antacids can reduce the absorption of so many other drugs, including phenytoin, several β-blockers, isoniazid, and digoxin, that as a general rule, patients should be advised to stagger the dosing of antacids and other prescription drugs by a period of at least 2 h.
Food Effects Food can affect drug absorption through effects on gastrointestinal physiology and through physical effects on drugs in the lumen. The magnitude and direction of a food effect depends on the specific characteristics of the meal, including the composition of the meal (proteins, carbohydrates, or fats), calorie content, and the total volume of food and fluid ingested. Physiological changes brought about by food include a delay in gastric emptying; changes in gastrointestinal pH, stimulation of bile flow, and increased liver blood flow, all of which can affect drug absorption. Absorption can also be affected by the physical presence of food in the gastrointestinal lumen. Food can impede drug diffusion to the membrane and reduce absorption; and constituents in food, such as ions, can chelate drugs and reduce absorption. Increased gastric residence time may increase dissolution in gastric fluid but can also increase the destruction of acid-labile drugs. As a general rule, the delay in gastric emptying brought about by food delays the absorption of most drugs because it slows their delivery to the small intestine, which is where most drugs are absorbed. The effect of food on the extent of absorption is more variable. A classification system in which drugs are categorized according to their permeability and dissolution characteristics has been used to help predict whether food will increase, decrease, or not affect the absorption of individual drugs (see Section 3.7).
3.5.5 Transcellular Passive Diffusion
Most drugs penetrate the gastrointestinal membrane by transcellular passive diffusion, driven by the concentration gradient across the membrane. As discussed in Chapter 2, the small intestine (duodenum, jejunum, and ileum) has an extremely large absorptive area, which results in it being the primary site for drug absorption. The small intestine is also highly perfused with blood. This allows absorbed drug to be rapidly carried away from the absorption site, and enables a high concentration gradient to be maintained across the intestinal membrane. Because of the more extensive absorption of the drug from the intestine, the rate of stomach emptying plays an important role in controlling the speed with which drugs are absorbed. Slow stomach emptying delays the delivery of drug to the primary site of absorption and slows down absorption, whereas rapid stomach emptying can speed up drug absorption.
A drug’s transcellular permeability is determined primarily by its lipophilicity, which as discussed in Chapter 2, can be quantified using the drug’s partition coefficient (P) or distribution coefficient (D). Drugs with negative log P or log D values, indicative of high hydrophilicity and poor lipophilicity, cannot easily penetrate the lipid matrix of a membrane. Conversely, drugs with high log P or log D values generally have good permeability. A study of almost 500 therapeutic drugs found that 50% had log P > 2 and 80% had log P > 0 (2). However, compounds with high log P or log D values are also most likely to have poor dissolution in aqueous media (5). As a result, values of log D7.4 in the range of about 1 to 3 are considered optimum for balancing permeability and dissolution. The size of a drug molecule is also important. Large molecules such as peptides and proteins are unable to penetrate the intestinal membrane. Drugs with a molecular mass greater than 500 Da are predicted to have poor membrane permeability, particularly if they also possess additional adverse physicochemical characteristics, such as polarity or low lipophilicity (6). Studies indicate that for drugs with molecular masses below around 400 Da, size is not an important factor for membrane penetration (7). Under these circumstances, a drug’s log P or log D value at a physiological pH, combined with a measure of the drug’s polarity (polar surface area or number of hydrogen bond donors), can be used to predict transcellular passive diffusion.
3.5.6 Paracellular Passive Diffusion
The junctions between the endothelial cells of the gastrointestinal membrane are very tight, which makes it extremely difficult for all but very small drugs to penetrate. Furthermore, the gaps between adjacent cells in the small intestine constitute only a very small fraction (0.01 %) of the total absorptive area, which makes it a very unappealing and inefficient route of drug penetration (2). Small polar drugs such as atenolol (MW 266 Da; log D7.4 ≈ -1.5) were believed to be absorbed by this route (5). However, recent work has cast doubt on this and suggests that the enormous absorptive area available for transcellular absorption may enable small hydrophilic drugs such as atenolol to undergo some transcellular absorption (2). The aminoglycoside antibiotics, which are both very polar (log D7.4 ≈ -10) and very large (MW 450 to >1000 Da), are unable to penetrate the intestinal membrane by either route. These drugs must be given parenterally and are usually administered as short, intermittent intravenous infusions for the treatment of systemic infections.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


