Fig. 3.1
A schematic diagram showing the enteral routes of drug administration (oral, sublingual and rectal) and their relative susceptibility to first-pass elimination, influencing the absorption and bioavailability of a drug. Following oral administration, Drug A is 100 % absorbed but 40 % bioavailable, while Drug B is 80 % absorbed and 60 % bioavailable. Hepatic first-pass elimination (1 – Fractional bioavailability) of Drug A is 60 % while that of Drug B is 40 %. Following sublingual administration, Drug A is 100 % bioavailable while following rectal administration, Drug A bioavailability is between 40 and 100 %, depending how far up the rectum the drug product is inserted
Measurement of Bioavailability
For intravenous injection, the whole of drug dose is delivered directly into the systemic circulation, thus it will always have 100 % bioavailability (F = 1) and the maximal plasma concentration (Cmax) is reached instantaneously (Tmax = 0 min). For other parenteral routes such as subcutaneous and intramuscular injections, the bioavailability may still be close to 100 % for most therapeutic drugs (F = 0.75−1), since little or no significant metabolism of the drug occurs in the skin or muscle and these small molecules (MW <800) would have no problem permeating the capillary endothelium, but the time to reach its maximum plasma concentration may be relatively slower (Tmax > 0 min) compared to the intravenous route. As for orally administered drugs, their bioavailabilities are often below 100 % (F < 1) because of incomplete absorption and/or first-pass elimination. Their Cmax and Tmax may also differ for the different dosage forms (Fig. 3.2).
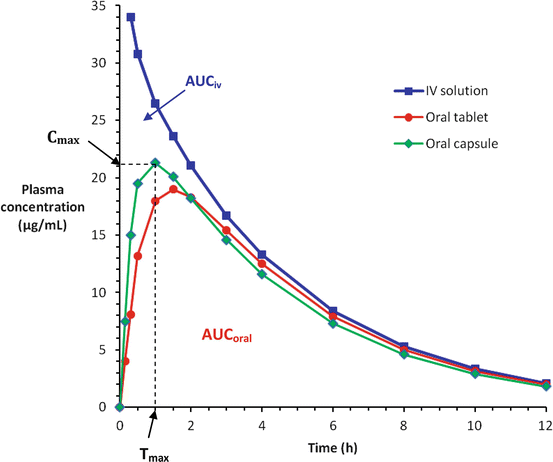

Fig. 3.2
Plasma concentration-time curves for a drug given by intravenous (IV) injection and two oral dosage forms (tablet and capsule). Cmax = Peak concentration; Tmax = Time-to-peak concentration; the oral bioavailability is determined by F = AUCoral/AUCiv, where AUC = area under the plasma concentration-time curve for the respective routes
Clinical Relevance of Bioavailability
A low systemic bioavailability will obviously result in the amount of drugs reaching the target tissue being reduced yielding a smaller than expected drug response. If the bioavailability is so poor, it is possible that the administered drug dose may not even reach the minimal effective concentration to produce the desired clinical effect. To overcome this, we can increase the dose administered, change the pharmaceutical formulation, or use a different route of administration.
Drug Permeation
During the process of absorption a drug must cross biological barriers in order to get from the site of administration into bloodstream. There are four major mechanisms by which drug molecules permeate cell membranes: diffusion through lipid, diffusion through aqueous channels, carrier-mediated transports (both active transport and facilitated diffusion) and pinocytosis. Of these mechanisms, diffusion through lipid and carrier-mediated transport are the ones most commonly encountered in drug absorption. The aqueous channels (about 0.4 nm in diameter) are too small to allow most drug molecules (usually >1 nm diameter) to pass through. Pinocytosis appears to be important for the transport of some big molecules (e.g., insulin across the blood brain barrier), but not for small molecules like most drugs.
Diffusion Through Lipid
Non-polar drug molecules passively diffuse across the membrane lipid according to its permeability coefficient, P, and the concentration difference across the membrane. Two physicochemical factors contribute to P, namely the solubility of the drug in the membrane lipid and its diffusivity. Lipid diffusion is by far the most important mechanism by which drugs cross intestinal mucosa to enter portal circulation.
Carrier-Mediated Transport
A number of lipid-insoluble drugs (e.g., levodopa, fluorouracil, iron and calcium) resemble endogenous substances and are carried across cell membranes by forming complexes with specific transmembrane proteins called carriers or transporters. This carrier-mediated transport may operate purely passively (as in facilitated diffusion) or it may be coupled to the electrochemical gradient of Na+ (as in active transport). Carriers are proteins and like receptors, they exhibit selectivity and saturability, and are also subject to competitive inhibition. In addition, active transport may be blocked by inhibitors of cellular metabolism.
Factors Affecting Drug Absorption and Bioavailability
Oral Route
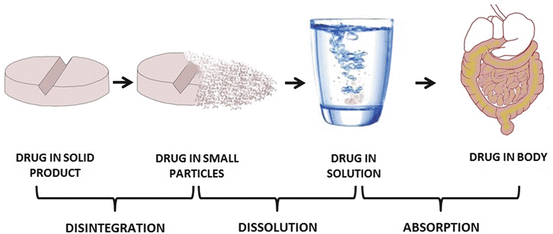
In most instances, absorption occurs when drug molecules are in the form of solutes. If a drug is given in solid form, then the drug must first break down into smaller particles (disintegration) and dissolve in the medium in which it is administered (dissolution) before it can traverse the cell membrane (permeation) and enter the bloodstream (absorption), Fig. 3.3. Thus the rate at which a given medication is absorbed will depend on the relative speed at which these processes occur, and the overall rate of absorption is often determined by the step with the slowest rate. Except for controlled-release medication, disintegration of a solid drug product usually occurs more rapidly than drug dissolution and permeation. Thus, in general, drug absorption may be dissolution rate-limited or permeation rate-limited.