Dose-response Relationships in Clinical Trials
There is no more difficult art to acquire than the art of observation, and for some men it is quite as difficult to record an observation in brief and plain language…. Observe, record, tabulate, communicate. Use your five senses…. Learn to see, learn to hear, learn to feel, learn to smell and know that by practice alone you can become expert.
–Sir William Osler
INTRODUCTION
In the past, most pharmaceutical companies were not overly concerned about exploring the entire dose-response range for each of their investigational drugs. They were primarily interested in finding a reasonably safe dose at which appropriate clinical efficacy could be demonstrated, and the dose was then doubled, tripled, or more substantially increased to obtain an assessment of the drug’s relative safety. If the tested doses were on the plateau phase of the dose-response relationship for the drug’s efficacy, a flat dose-response curve was observed, and companies then felt confident in recommending the range of doses tested. If tested doses were on the steep part of the dose-response relationship, the peak effect may or may not have been observed with the highest dose tested. This approach usually did not find the lowest part of the dose-response relationship.
Prior to the 1990s, few companies were required by regulatory authorities to explore the entire dose-response relationship for either safety or efficacy. As a result, numerous drugs were marketed, often for many years, at doses that were higher than necessary. For example, captopril and thiazides were prescribed for hypertensive patients at doses later found to be much greater than therapeutically necessary for most patients.
The dose-response relationship for both individuals and groups of patients has received significant academic attention (Lasagna, Erill, and Naranjo 1989), and regulatory authorities are also focusing attention on this issue (Temple 1989). More and more regulatory reviewers believe that a company should have at least some information on the entire dose-response relationship at the time of a drug’s initial marketing. In the opinion of regulators, special patient populations that react differently to the drug should also be evaluated to see if they have a different dose-response relationship than the “general” population. These special populations include patients of a particular age, with impaired physiological function, or with important risk factors.
Considerations of Dose
The dose of a drug is the precise amount given to a patient or to a specific part of the patient (e.g., in an isolated limb perfusion to treat certain tumors). The dose is not always an accurate indication of either the concentration of the drug in the plasma or the amount that reaches receptors. Factors such as particle size of the active ingredient and what excipients are combined with the active ingredient may be critical to understanding why a specific dose is poorly absorbed or causes a much greater or lesser effect than anticipated. Nonetheless, the dose of a drug is usually related to the plasma concentration and amounts that reach receptors.
Considerations of Response
In a clinical trial, the net clinical response is either a reaction that is positive (i.e., potentially or actually beneficial to the patient) or negative (i.e., potentially or actually harmful to the patient). The response is a complex summation of numerous events, including:
The natural progression of disease
Influence of nondrug factors
Influence of concomitant drugs
Influence of concomitant nondrug treatments
Placebo response to the trial drug
Biological effects of the trial drug
Each of these can lead to improvement and/or deterioration of the patient. This makes the overall response a complex summation and one that is often impossible to predict in advance with any certainty.
Some negative responses (e.g., adverse events) may be shown to be positive for other patients. For example, unwanted hair growth was observed in women as an adverse event of minoxidil but was eventually viewed as a potential positive response in balding men. Subsequent clinical trials established the drug’s efficacy in growing scalp hair in some bald men. Another example concerns the unwanted sedation caused by some antihistamines that was turned into a potential benefit when one of these drugs was marketed as an aid to promote sleep. Thus, clinical responses are not always either purely beneficial or purely deleterious.
Dose-response-time Relationships
Dose-response relationships are really two axes of a three-dimensional relationship. The third dimension is time. Time measures the latent period before a drug starts to act, the duration of a drug’s effect, and the duration of dosing required [e.g., treatments may either be continuous (e.g., with infusions) or intermittent (e.g., oral dosing)]. Time may be measured in terms of years (e.g., to determine when booster injections are required for certain vaccines). Time is also a major factor in assessing the onset and the plateau effect of efficacy of antidementia and many other drugs used in psychiatry. This is particularly true when it takes a number of months to see a clear beneficial effect. Thus, dose-response evaluations are difficult to conduct in these situations.
While a trial may have three or more groups given fixed doses, intersubject variability suggests that, often, more dose-response information will be obtained by titrating individual patients and obtaining individual dose responses. However, when the onset of activity is several months, it is not easy to conduct those trials, and one usually reverts to the former method. In the field of anesthesia, however, the rapid response to most drugs facilitates the evaluation of dose-response relationships during clinical trials.
Goals of Dose-response Trials
A major goal of dose-response trials is to identify the optimal dose or dose range appropriate for a specific indication in terms of both safety and efficacy for a specific population of patients. This involves determining the:
Amount of drug to be taken in each dose
Number of doses to be taken each day
When during the day (or night) each dose is to be taken
Conditions under which each dose is to be taken (e.g., with food, the amount of water)
As the dose given to each patient or group of patients increases in a clinical trial, the response of each patient may also increase (i.e., a greater effect is observed). In addition (or instead), more people may respond to treatment. The second case is more likely to occur if clinical responses are not graded but, instead, are all-or-none.
Goals of Dose-response Determinations
The major reason for conducting dose-response trials at the individual patient level is to learn how to treat optimally specific types of patients. The objective is to provide, to individual patients, one or more drugs with the greatest degree of safety, at the least cost, and with the fewest clinically significant adverse events and interactions. Another objective is to provide regulatory authorities with sufficient data to convince them that physicians can adequately control the drug in their patients.
When Are Dose-response Relationships Usually Determined during New Drug Development?
Dose-response relationships are explored during all three premarketing phases of drug development. During Phase 1, an ascending dose-response trial is conducted to assess safety in cohorts of normal volunteers or in seriously ill or refractory patients (e.g., for anticancer drugs). When these trials are conducted in the United States, it is traditional to explore the
dose-response relationship over the entire dose range (i.e., until unacceptable adverse events occur). This is initially done by evaluating each dose in a single-dose volunteer (or patient) trial and then, in a second or third trial, evaluating multiple doses, usually over several days to a week, in a volunteer (or patient). In some European countries, doses are typically explored only up to those amounts where plasma level concentrations are achieved that are expected to yield efficacy in Phase 2 trials.
dose-response relationship over the entire dose range (i.e., until unacceptable adverse events occur). This is initially done by evaluating each dose in a single-dose volunteer (or patient) trial and then, in a second or third trial, evaluating multiple doses, usually over several days to a week, in a volunteer (or patient). In some European countries, doses are typically explored only up to those amounts where plasma level concentrations are achieved that are expected to yield efficacy in Phase 2 trials.
Phase 2 pilot trials usually evaluate two or more doses of a drug to seek efficacious effects. Phase 2 well-controlled trials often include at least two doses of the test drug. Phase 3 trials often involve dose-response evaluations in special populations, or they may involve evaluations of two or more doses given over longer periods in populations of subjects taking other concomitant drugs and/or who have concurrent illnesses.
TYPES OF DOSE-RESPONSE TRIALS
Dose-ranging Trials
These are open-label or double-blind pilot trials that are specifically designed to evaluate the safety and/or preliminary efficacy of a number of doses. Dose-ranging trials may be conducted by titrating individual subjects or by giving fixed doses to two or more groups to study their effects. Essentially, the dose-ranging trial is a dose-response trial but at an early stage of development when one is seeking to learn the active dose of a drug, an inactive dose, and the peak dose that yields a plateau effect of efficacy but is still acceptably safe for treating patients.
Pharmacokinetic Trials
These Phase 1 trials (usually open-label) often compare pharmacokinetic parameters (e.g., absorption, peak concentration, half-life) obtained with two or more doses of a drug. More sophisticated pharmacokinetic trials conducted when a drug enters Phase 3a explore the pharmacokinetics of special populations (e.g., elderly, pediatric, immunocompromised, those with particular organ failures) and also explore specific areas (e.g., interactions with various types of diets, drug-drug interactions).
Metabolic Trials
Mass balance and metabolic trials may be designed to evaluate a drug’s metabolic rate and may evaluate multiple doses. Although, strictly speaking, metabolic trials are part of the field of pharmacokinetics, they are often designed by someone other than a pharmacokineticist.
Dose Tolerance Trials
In dose tolerance trials, patients or healthy volunteers are exposed to the maximal amount(s) of a single dose or multiple doses that they can tolerate. This procedure is usually, but not always, conducted as part of a Phase 1 trial. It provides information about the top of the safety dose-response relationship.
Double-blind Trials Evaluating Two or More Doses or Dose Ranges
These trials can be designed by titrating doses in all patients, either individually or as a group, to reach predetermined doses in Phase 2 or 3. These evaluations are also sometimes conducted as part of Phase 4 trials.
There are some occasions when dose-response relationships should not be determined. It is usually inappropriate to determine dose-response relationships for efficacy of chemotherapeutic drugs that treat neoplastic or invasive diseases. This is because these drugs are usually dosed to reach maximal levels that are tolerated. In the field of antibiotics, when submaximal doses are given to patients, there is an increased probability of bacterial resistance developing because of the variation in sensitivity of the organism to the drug. For example, most patients may be adequately treated for pneumococcal pneumonia with about 100,000 units of penicillin per day. Instead, patients are treated with 2,400,000 units per day because of varying sensitivity of the organisms to the drug and the need to ensure that all bacteria are killed. A later section in this chapter describes a few other situations when it is inappropriate to obtain dose-response relationships in clinical trials (e.g., to evaluate allergic responses or irreversible drug effects).
ESTABLISHING DOSE-RESPONSE RELATIONSHIPS IN CLINICAL TRIALS
The dose-response relationship for safety is usually evaluated in the first Phase 1 clinical trial and involves a stepwise examination of safety beginning with the lowest dose tested. A single dose is given to a cohort of volunteers or patients, and after safety is assessed and found to be acceptable, an increased dose is given to the second cohort of volunteers or patients. This is an example of a bottom-up approach to developing a dose-response relationship, which is usually most appropriate when safety is the primary objective of the clinical trial or when the drug is known to have a narrow therapeutic window.
It is possible to develop a dose-response relationship for efficacy in a clinical trial starting from the threshold, midpoint, or upper part of the safety dose-response relationship. It is usually faster (and more appropriate), however, to define the entire efficacy dose-response relationship by starting at the upper end of the dose that is tolerated and titrating down when subjects are unable to tolerate the dose. This is referred to as the top-down approach to establishing a dose response. The top-down approach is inappropriate for establishing a dose-response relationship for safety. These two approaches are described in more detail in the following sections.
Top-down Approach to Establishing a Dose-response Relationship
In this approach, it is important to establish, in Phase 1 trials, the highest dose of a drug that can be tolerated by normal volunteers. This is accomplished by first determining (if possible) the highest single dose tolerated by normal volunteers. Patients, rather than volunteers, are enrolled in Phase 1 trials of potential new drugs for cancer, anti-acquired immunodeficiency disorder (AIDS) agents, antiarrhythmics, and a few other indications where the drug is known or believed to be toxic or where the therapeutic window is believed to be narrow.
A second clinical trial using multiple doses generally given for two to seven days is then conducted. This trial usually demonstrates that volunteers (or patients) can tolerate a lesser maximal dose when multiple doses of the drug are given. At that point, the highest tolerated dose is generally able to be identified. Patients are then given doses that are titrated up to the highest tolerated dose in one or more Phase 2 dose-ranging trials to evaluate their tolerance and the efficacy of several doses of the drug.
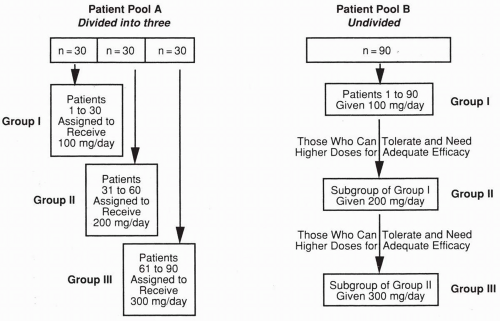 Figure 63.1 Patient pools that illustrate two approaches to a dose-response evaluation. See text for details. |
For most drugs, patients tolerate a lower dose than volunteers. This means that the dose-ranging trial design evaluates both efficacy and safety at the top of the dose-response relationship for safety that was determined in Phase 1 trials. If patients are unable to tolerate the maximal dose intended to be used in the Phase 2 trial, the dose is progressively lowered patient-by-patient until a dose is found that is well tolerated by most patients. The range of peak-tolerated doses establishes the top of the dose-response relationship that may be studied in additional trials. Other clinical trials are conducted at lower doses to explore the lower part of the dose-response relationship, including the general range of threshold doses for establishing efficacy.
For some drugs (e.g., benzodiazepines), patients are able to tolerate higher doses than volunteers and require higher doses than expected based on animal data. This finding may or may not be expected. If it is unsuspected prior to Phase 2, then efficacy may not be observed in a conservatively designed pilot dose-ranging trial. A drug that is extremely safe and does not elicit adverse events at doses tested in Phase 1 volunteers may eventually be found also to be ineffective or not optimally effective in Phase 2. In such cases, it may be necessary to repeat a Phase 1 trial using higher doses than previously tested prior to testing some (or all) of those higher doses in a new Phase 2 trial.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


