Chapter 11 DNA Technology
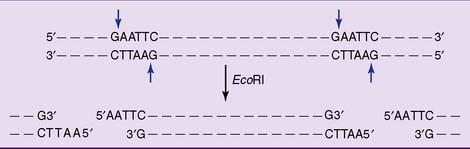
Restriction endonucleases cut large DNA molecules into smaller fragments
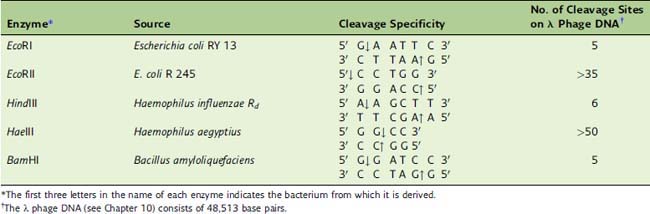
Most restriction enzymes make staggered cuts one or two base pairs away from the symmetry axis of their recognition sequence in both strands. Therefore the double-stranded restriction fragments have short single-stranded ends (Fig. 11.1 and Table 11.1).
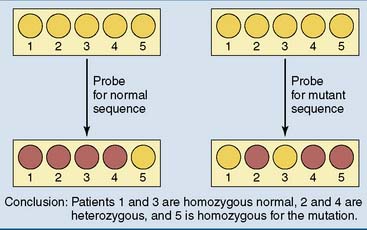
Dot blotting is used for genetic screening
Dot blotting (Fig. 11.2) is a rapid, inexpensive screening test for the detection of small mutations and polymorphisms. The extracted and denatured DNA is applied to two strips of nitrocellulose paper, which binds the single-stranded DNA tightly. One strip is dipped into a solution containing an oligonucleotide probe for the normal sequence, and the other is dipped into a solution with a probe for the mutation. If only the probe for the normal sequence binds, the patient is homozygous normal. If only the probe for the mutation binds, the patient is homozygous for the mutation. If both probes bind, the patient is heterozygous.
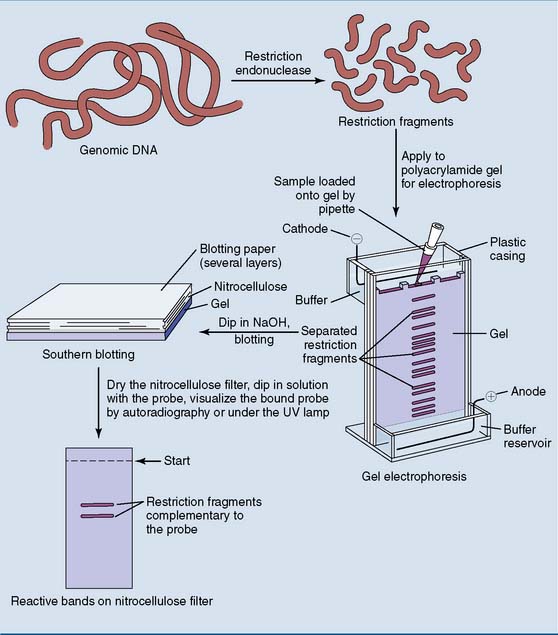
Southern blotting determines the size of restriction fragments
As shown in Figure 11.3, restriction fragments obtained from genomic DNA are separated by electrophoresis in a cross-linked agarose or polyacrylamide gel. This method separates the restriction fragments by their size rather than their charge/mass ratio. Small fragments move fast, and large fragments move slowly because they are retarded by the gel.
DNA can be amplified with the polymerase chain reaction
The procedure (shown in Figure 11.4) uses a heat-stable DNA polymerase such as Taq polymerase. This enzyme is derived from Thermus aquaticus, a thermophilic bacterium that was originally isolated from a hot spring in Yellowstone National Park. It functions best at temperatures close to 60°C and can survive repeated heating to 90°C.
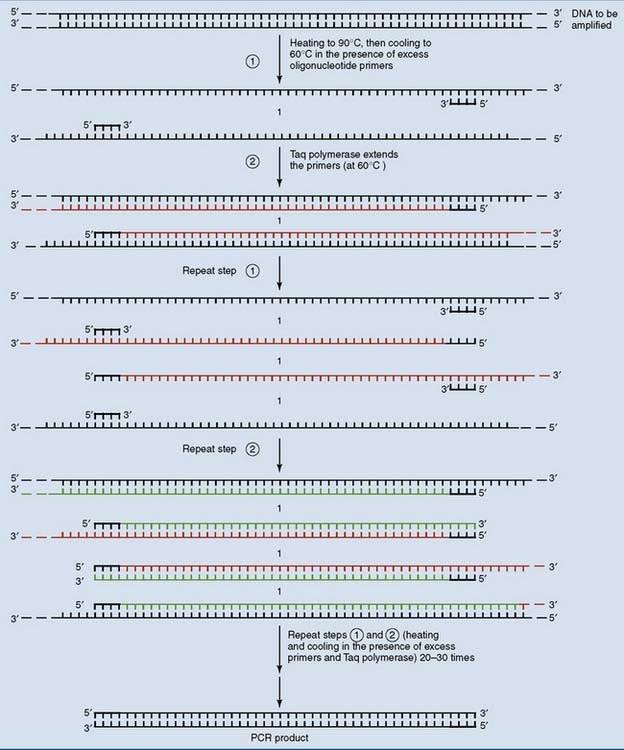
 , Oligonucleotide primer.
, Oligonucleotide primer.PCR is used for preimplantation genetic diagnosis
Figure 11.5 shows the use of PCR with nested primers for preimplantation diagnosis of the ΔPhe508 mutation. This three–base-pair deletion is readily identified by PCR followed by gel electrophoresis because the mutated sequence yields a PCR product three base pairs shorter than normal. However, base substitutions cannot be identified by electrophoresis alone. They require the application of allele-specific probes to the PCR product.
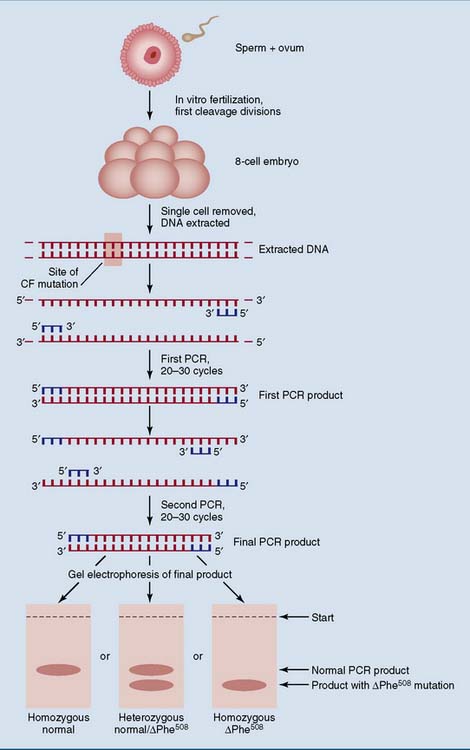
Figure 11.6 shows how these deletions are identified by amplification of deletion-prone exons. If one of the target exons is deleted, its PCR product is absent.
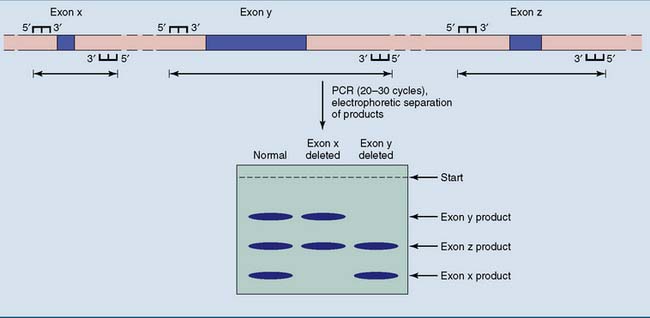
 , Exons;
, Exons;  , primer.
, primer.Allelic heterogeneity is the greatest challenge for molecular genetic diagnosis
Normal polymorphisms are used as genetic markers
Microsatellite polymorphisms are even more useful. These are tandemly repeated sequences with, in most cases, two to four nucleotides in the repeat unit and a total length well below 1000 base pairs (Fig. 11.7, B). The number of repeat units, and therefore the length of the microsatellite, varies among people. Whereas SNPs and RSPs have only two alleles, the most useful microsatellites have more than two alleles. Polymorphic microsatellites produce restriction fragments and PCR products of different length that can be separated by gel electrophoresis.
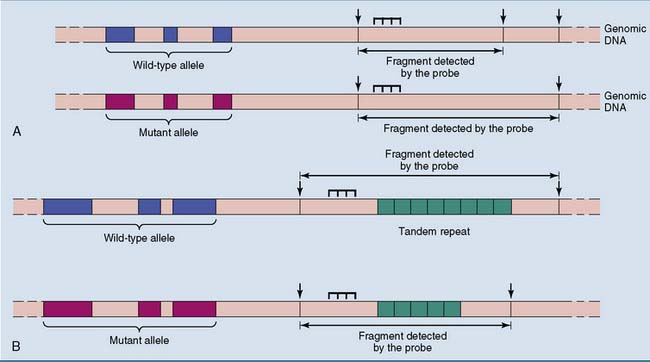
 , probe used to detect the polymorphism.
, probe used to detect the polymorphism.Tandem repeats are used for DNA fingerprinting
DNA fingerprinting can be performed with Southern blotting or PCR (Fig. 11.8). Southern blotting requires a substantial amount of DNA, for example, from a drop of seminal fluid from a sex offender. PCR is used when only a small amount of DNA is available, for example, from a single hair of the murderer stuck under the victim’s fingernail. However, because of its high sensitivity, PCR is more vulnerable to contamination by extraneous DNA. This could put the laboratory technician at risk for being wrongly convicted!
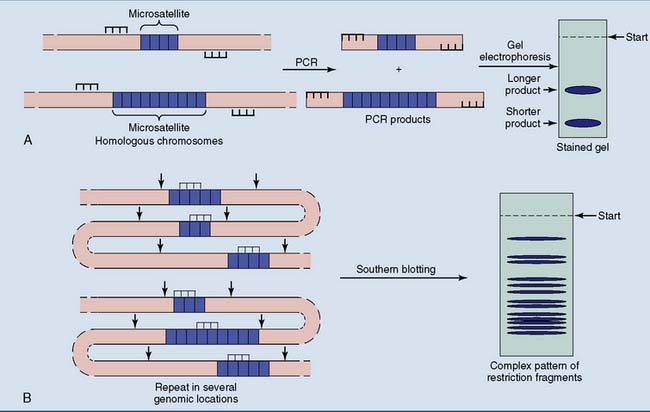
 , Primer;
, Primer;  , probe; ↓, restriction site.
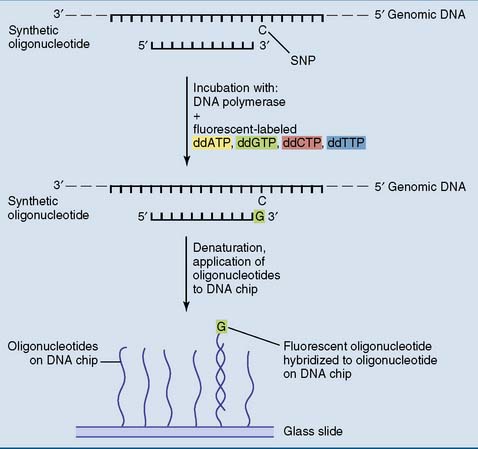
, probe; ↓, restriction site.DNA microarrays can be used for genetic screening
Fluorescent-labeled genomic DNA fragments that are applied to the microarray hybridize primarily with the exact complementary probe but not the probe for the alternative allele. For example, when one square of the microarray has a probe for the sickle cell mutation and another has a probe for the corresponding normal sequence, normal DNA produces substantially more fluorescence on the square for the normal sequence. DNA from a sickle cell patient produces substantially more fluorescence on the square with the probe for the sickle cell mutation, and the DNA of a heterozygote produces about equal fluorescence on both. However, more imaginative methods are being explored as well (Fig. 11.9).