Introduction
Male reproductive tract functions include androgen homeostasis, spermatogenesis, sperm transport and storage, and normal erectile and ejaculatory function ability. The control of these functions involves the pituitary gland, central and peripheral nervous systems, and genitalia. In addition to a review of normal male reproductive anatomy and physiology, this chapter considers two common disorders of the male reproductive tract: male infertility and benign prostatic hyperplasia.
Normal Structure & Function of the Male Reproductive Tract
The male reproductive tract is composed of the testes, genital ducts, accessory glands, and penis (Figure 23–1).
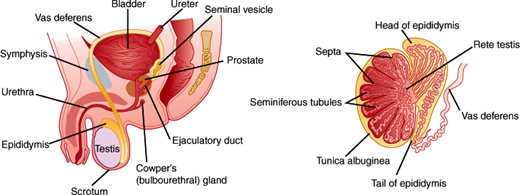
The testes are responsible for the production of testosterone and spermatozoa. Each testis is approximately 4 cm in length and 20 mL in volume. The testis is divided into lobules consisting of seminiferous tubules (inside which sperm are produced) and connective tissue (Figure 23–2). The seminiferous tubules converge to form another network of tubules called the rete testis through which spermatozoa are transported to the epididymis.
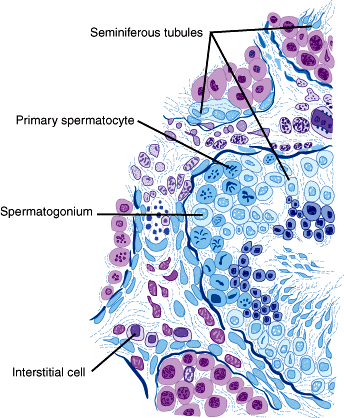
The seminiferous tubules are surrounded by a basal membrane and a specialized epithelium containing Sertoli cells that provide protection and nourishment to germ cells. Commensurate with puberty, tight junctions develop between adjacent Sertoli cells, creating an impermeable lining called the blood-testis barrier. This barrier divides the seminiferous tubules into a basal compartment and an adluminal compartment, separating more advanced germ cells from the immune system. The separation is necessary because mature sperm are potentially antigenic since they are not present at the prepubertal interval when much of the immune tolerance is established. The Leydig cells in the intertubular connective tissue produce testosterone.
Both testosterone production and spermatogenesis are controlled by the hypothalamic-pituitary-gonadal axis. The hypothalamus produces gonadotropin-releasing hormone (GnRH) in a pulsatile fashion. GnRH courses through the hypothalamic-pituitary portal system to stimulate the anterior pituitary gonadotropes to secrete (also in a pulsatile fashion) the two gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH). FSH stimulates the Sertoli cells to produce paracrine growth factors and other products supporting spermatogenesis. FSH also stimulates the production of inhibin in response to active spermatogenesis and androgen-binding globulin (ABP).
Under the influence of LH, the Leydig cells produce testosterone. Concentrations of testosterone in the seminiferous tubules are 80–100 times greater than in the general circulation. Androgens act on spermatogenesis via the Sertoli cells, and high testicular levels of androgens are essential for spermatogenesis. Circulating testosterone provides negative feedback on secretion of GnRH, LH, and FSH by acting on both the hypothalamus and the pituitary. Gonadal inhibin exerts negative feedback on FSH secretion by the pituitary.
During spermatogenesis, primitive germ cells develop into mature spermatozoa while moving from the basement membrane to the lumen of the tubules. The immature germ cells near the basement membrane are called spermatogonia and have the normal diploid number of 46 chromosomes. Beginning at puberty and continuing throughout life, the spermatogonia divide mitotically, maintaining the population. Some of the spermatogonia differentiate into primary spermatocytes and enter the first meiotic division. During the prophase of the first meiotic division, duplication of DNA, pairing of homologous chromosomes, and crossing over take place in the spermatocytes as they develop a duplicated set of 46 chromosomes. The spermatocytes (now called secondary spermatocytes) then undergo the second meiotic division producing spermatids, which have a haploid number of unduplicated chromosomes. In this way, four spermatids are produced from each spermatogonium. Spermatids then undergo a maturation process called spermiogenesis to form spermatozoa. In this process, condensation of the nuclear chromatin takes place and the enzyme-filled acrosome cap is formed. The spermatids also elongate and develop flagella. Spermiogenesis ends with the spermatozoa being released from the germinal epithelium. The process in which the primary spermatogonia divide and develop into mature spermatozoa takes about 74 days.
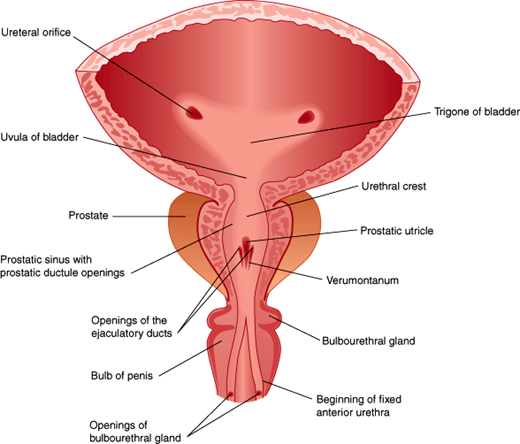
After spermiogenesis, spermatozoa are released into the lumen of the seminiferous tubule, then course through the rete testis into the epididymis. During a 5- to 14-day epididymal transit, spermatozoa mature and become capable of progressive movement in a process involving changes in membrane, metabolism, and morphology. Sperm are stored in the cauda epididymis until the time of ejaculation. During ejaculation, sperm travel through the vas deferens via the inguinal canal and medially to the posterior and inferior part of the urinary bladder where the vas deferens fuse with the duct of the seminal vesicle, forming the combined ejaculatory duct. The ejaculatory ducts enter the prostatic portion of the urethra at the verumontanum distal to the internal bladder sphincter (Figure 23–3).
During normal erection, parasympathetic fibers travel from S2-S4 through the pelvic nerve and the pelvic plexus to the cavernous nerve where they release acetylcholine (ACh) and nitric oxide (NO). Their release causes relaxation of the smooth muscles of the penile corpora, which in turn leads to increased blood flow and blood trapping, resulting in erection.
The ejaculatory reflex is initiated primarily by afferent input from the dorsal nerves of the penis, although cerebral erotic stimuli can also play a role. The ejaculate is transported from the ampulla of the vas deferens into the posterior urethra in a process called seminal emission. This is the result of peristaltic contractions of smooth muscle cells in the epididymis, vas deferens, and accessory sex glands under sympathetic control from fibers arising from T10-T12. After seminal emission, contraction of the posterior urethra and closure of the bladder neck (preventing retrograde ejaculation into the bladder) are initiated by sympathetic nerve fibers, while the external urethral sphincter relaxes. These events are followed by rhythmic contractions of the periurethral and pelvic floor muscles mediated by the somatic fibers from S2 to S4 running through the pudendal nerve. This results in the projectile phase of ejaculation.
In the female reproductive tract, the spermatozoa must migrate through the cervical mucus and then undergo a series of functional and structural changes collectively termed capacitation. These changes are necessary for the spermatozoa’s ability to fertilize the oocyte as they facilitate the acrosome reaction, during which the sperm plasma membrane fuses with the outer acrosomal membrane. This exposes the contents of the acrosome, such as acrosin and hyaluronidase, allowing penetration of the oocyte. Capacitation can also be induced by incubation in suitable laboratory medium.
Sperm cells only make up 1–2% of the semen volume, while the rest comes from the accessory male sex glands. The seminal vesicles produce two-thirds of the ejaculate volume and provide fructose as an energy source, as well as seminogellin, which contributes to seminal coagulation. The prostate supplies about one-third of the ejaculate, and this includes prostate-specific antigen, a proteolytic enzyme that cleaves seminogellin, effecting liquefaction. Finally, the bulbourethral glands contribute a small amount of clear mucoid discharge, released mainly during sexual stimulation before ejaculation.
The testes secrete two steroid hormones that are essential to male reproductive function: testosterone and dihydrotestosterone. The pathways for testicular androgen biosynthesis are illustrated in Figure 23–4.
Figure 23–4
Biosynthesis and metabolism of testosterone. Heavy arrows indicate major pathways. Circled numbers represent enzymes as follows:  cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1);
cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1);  hydroxy-Δ-5-steroid dehydrogenase, 3β- and steroid Δ-isomerase (HSD3β);
hydroxy-Δ-5-steroid dehydrogenase, 3β- and steroid Δ-isomerase (HSD3β);  17α-hydroxylase activity of cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1);
17α-hydroxylase activity of cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1);  17,20-lyase activity of cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1);
17,20-lyase activity of cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1);  hydroxysteroid (17β) dehydrogenase (HSD17β);
hydroxysteroid (17β) dehydrogenase (HSD17β);  steroid-5α- reductase, polypeptide 2 (3-oxo-5α-steroid Δ4-dehydrogenase α) (SRD5A);
steroid-5α- reductase, polypeptide 2 (3-oxo-5α-steroid Δ4-dehydrogenase α) (SRD5A);  cytochrome P450, family 19, subfamily A, polypeptide 1 (CYP19A1). (Modified and redrawn, with permission, from Gardner DG et al. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw Hill, 2011.)
cytochrome P450, family 19, subfamily A, polypeptide 1 (CYP19A1). (Modified and redrawn, with permission, from Gardner DG et al. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw Hill, 2011.)
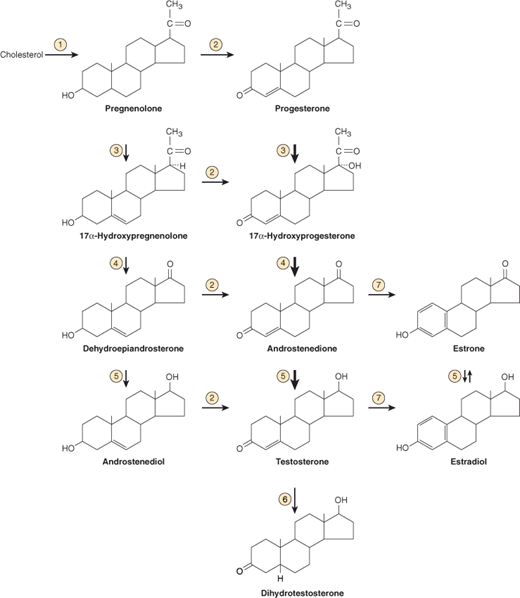
Testosterone, a C19 steroid, is synthesized from cholesterol by the interstitial (Leydig) cells of the testes and from androstenedione secreted by the adrenal cortex. The majority of circulating testosterone is bound to sex-hormone–binding globulin (SHBG) and is unavailable for biological activity.
The remainder is loosely bound to albumin and is available for target tissue action. Only about 2% is unbound in circulation. The albumin-bound and free fractions make up the “bioavailable” testosterone in circulation. SHBG is synthesized in the liver and may be increased in certain clinical conditions. The effect of increasing SHBG in circulation is to lower the bioavailable fraction, so that while the serum total testosterone level is normal, hypogonadism occurs at the tissue level because of protein binding. The most common causes of increased SHBG are liver dysfunction, hyperestrogenemia, obesity, and aging. Normal testosterone levels through the lifespan are characterized in Figure 23–5. The negative feedback control mechanisms for testosterone are depicted in Figure 23–6.
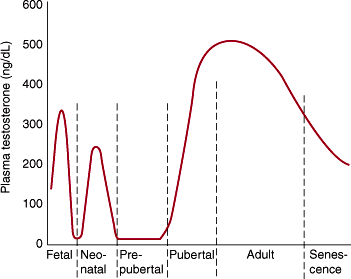
Figure 23–6
Endocrine control of the male reproductive system. ABP, androgen-binding protein; GnRH, gonadotropin-releasing hormone; T, testosterone; E2, estradiol; DHT, dihydrotestosterone. (Redrawn and modified, with permission, from Gardner DG et al. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw Hill, 2011.)
Dihydrotestosterone (DHT) is derived both from direct secretion by the testes (~20%) and from conversion in peripheral tissues (~80%) of testosterone and other sex steroid precursors secreted by the testes and adrenals. DHT circulates in the bloodstream. The normal plasma DHT level for the adult male is 27–75 ng/dL (0.9–2.6 nmol/L) (Table 23–1).
| Hormone | Conventional Units | SI Units |
|---|---|---|
| Testosterone, total | 260–1000 ng/dL | 9.0–34.7 nmol/L |
| Testosterone, free | 50–210 pg/mL | 173–729 pmol/L |
| Dihydrotestosterone | 27–75 ng/dL | 0.9–2.6 nmol/L |
| Androstenedione | 50–200 ng/dL | 1.7–6.9 nmol/L |
| Estradiol | 15–40 pg/mL | 55–150 pmol/L |
| Estrone | 15–65 pg/mL | 55.5–240 pmol/L |
| FSH | 2–15 mIU/mL | 2–15 U/L |
| LH | 2–15 mIU/mL | 2–15 units/L |
| PRL | 1.6–18.8 ng/mL | 0.07–0.8 nmol/L |
Estradiol is produced by aromatization of testosterone in the peripheral circulation. The aromatase enzyme is present in abundant amounts in fatty tissue. Thus, obesity can increase conversion of testosterone, with resultant hyperestrogenemia, downregulation of the hypothalamic-pituitary-gonadal axis, and hypogonadism.
Circulating testosterone or DHT crosses the membrane of the target cell and enters the cytoplasm. Testosterone may be converted to the more potent DHT inside the target cell. Testosterone or DHT binds to the androgen receptor, and the complex is then transported to the cell’s nucleus, where it binds to DNA and initiates mRNA synthesis. The resultant proteins synthesized account for the subsequent androgenic changes that occur (Figure 23–7).
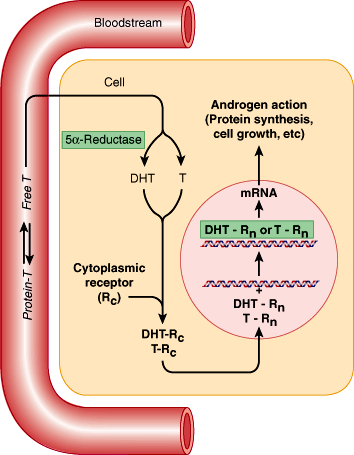
In the fetus, androgens are necessary for normal differentiation and development of the internal and external male genitalia. During puberty, androgens are needed for normal growth of the male genital structures, including the scrotum, epididymis, vas deferens, seminal vesicles, prostate, and penis. During adolescence, androgens and estrogens cause rapid growth of skeletal muscle and bone. Androgens are also responsible for development of the secondary sex characteristics summarized in Table 23–2. During adult life, androgens are necessary for normal male reproductive function. Specifically, androgens stimulate erythropoiesis, preserve bone structure and muscle mass, and maintain libido and erectile function.
| External genitalia | Penis increases in length and width; scrotum becomes pigmented and rugose |
| Internal genitalia | Seminal vesicles enlarge and secrete |
| Larynx | Larynx enlarges, vocal cords increase in length and thickness, voice deepens |
| Hair | Beard appears; scalp hairline recedes anterolaterally; pubic hair appears with male pattern (triangle with apex up); axillary, chest, and perianal hair appears |
| Musculoskeletal | Shoulders broaden; skeletal muscles enlarge |
| Skin | Sebaceous gland secretions increase and thicken |
| Mental | More aggressive, active attitude appears; libido develops |
Checkpoint
What is the purpose of seminiferous tubule tight junctions?
What are the roles of the two major cell populations in the testis, the Leydig cells and the Sertoli cells?
How is testosterone secretion regulated?
What are the target cells of luteinizing hormone (LH) and follicle-stimulating hormone (FSH)?
What are the relative concentrations of testosterone in the peripheral circulation and the testicular tissue?
Describe the sequence of events leading up to and during ejaculation.
How is estradiol created in men?
What are the effects of androgens?
Pathophysiology of Selected Male Reproductive Tract Disorders
For conception to occur, spermatogenesis must be normal and the seminal accessory glands must produce seminal fluids. The ducts for sperm transport must also be patent, and ejaculation must occur so the sperm can be delivered near the female’s cervix. Next, the spermatozoa must be able to travel to the uterine tubes, and they must undergo functional changes that allow them to fuse with the oolemma (plasma membrane of oocyte). Any defect in these mechanisms can result in infertility.
Infertility is defined as the inability of a couple to achieve pregnancy despite unprotected intercourse for a period of more than 12 months. About 15% of all couples are infertile, and it is estimated that a male factor plays a role in about half of the cases. In spite of this, the evaluation of the male partner is often neglected, mainly because of the high pregnancy rates that can be achieved by assisted reproductive techniques (ARTs). This practice is unfortunate because male infertility can often be cured, sparing the female partner the extensive treatment and cost of ART. Furthermore, evidence suggests that ART procedures can be associated with increased risks for both mother and child. Finally, neglecting to examine the infertile man properly risks overlooking serious conditions such as testicular cancer that may coexist with infertility.
Male infertility can be divided into pretesticular, testicular, and post-testicular forms. A comprehensive list of etiologies is presented in Table 23–3, genetic male infertility causes are listed in Table 23–4, and causes of testicular atrophy are in Table 23–5.
| Pretesticular | Testicular | Posttesticular |
|---|---|---|
|
|
|
| Disorder | Cause of Infertility | Defect |
|---|---|---|
| Chromosomal | ||
| Klinefelter syndrome | Oligoazoospermia, hyalinization of seminiferous tubules |
|
| XX male syndrome | SCOS |
|
| XYY male syndrome |
| |
| Genetic | ||
| Disorders of GnRH secretion | ||
| ||
| ||
| Decreased GnRH secretion |
|
| Defects in G protein coupled for GnRH |
|
| Decreased GnRH secretion |
|
| Disorders of androgen function | ||
| Excessive androgens inhibit pituitary secretion of gonadotropins |
|
| Androgen insensitivity syndromes (Reifenstein syndrome, testicular feminization, Lub syndrome, Rosewater syndrome) | Androgen insensitivity |
|
| Kennedy syndrome |
| |
| 5α-reductase deficiency |
| |
| Y chromosome microdeletions | ||
|
| |
| SCOS | |
| Variable phenotype: oligozoospermia to SCOS | |
|
| |
| Spermatogenic arrest | |
| Variable phenotype: oligozoospermia to SCOS | |
|
| |
| Variable phenotype: oligozoospermia to SCOS | |
| Azoospermia | |
| Congenital absence of vas deferens |
|
| Trauma |
| Testicular torsion |
| Hypopituitarism |
| Cryptorchidism |
| Klinefelter syndrome (47,XXY) |
| Alcoholism and cirrhosis |
| Infection (eg, mumps orchitis, gonococcal epididymitis) |
| Malnutrition and cachexia |
| Radiation |
| Obstruction to outflow of semen |
| Aging |
| Drugs (eg, estrogen therapy for prostatic cancer) |
The pretesticular causes of infertility originate in either the hypothalamus (GnRH) or the pituitary (LH and FSH). These endocrinopathies are most often caused by mutations in genes involved in the biosynthesis of the hormones, growth factors or receptors, and associated signal transduction pathways. The deficiencies result in a loss of intratesticular testosterone production and cessation of spermatogenesis.
Hypogonadotropic hypogonadism is an uncommon cause of male infertility but is important to recognize because replacement therapy can be initiated. The condition is characterized either by reduced GnRH production, causing circulating levels of FSH and LH to diminish, or by rare disorders of the pituitary (with normal GnRH) that result in primary deficiencies of FSH and/or LH. These defects result in deficient androgen secretion and spermatogenesis.
Disorders resulting in abnormal synthesis and release of GnRH are most often caused by mutations, small deletions, or polymorphic expansions within genes involved in the regulation of sexual development and function. Disorders of GnRH synthesis and release can also be caused by hypothalamic tumors. Disorders without a known cause are termed idiopathic hypogonadotropic hypogonadism. Men suffering from GnRH deficiencies have firm prepubertal-sized testes and a small penis.
Kallmann syndrome refers to a syndrome of defective olfaction with hypogonadotropic hypogonadism caused by failed olfactory and GnRH axonal migration during fetal development. This is caused by a mutation in the KALIG1 gene on Xp22.3 and results in deficiency in GnRH secretion and consequent failure of pubertal initiation along with anosmia or hyposmia. In addition, patients tend to be tall and can have congenital deafness, asymmetry of the cranium and face, cleft palate, cerebellar dysfunction, cryptorchidism, or renal abnormalities. However, some Kallmann syndrome patients present only with isolated gonadotropin deficiency, manifesting as infertility.
Other causes of pubertal failure include mutations in the recently discovered hypothalamic kisspeptin peptide or its cognate receptor GPR54. With clinical implications for diagnosis and treatment of infertility and related disorders, this ligand/receptor pair has proven to be one of the key mediators of pubertal onset.
Mutations of the X-linked Dax1 gene are associated with hypogonadotropic hypogonadism and congenital adrenal hypoplasia. Dax1 is a nuclear receptor that plays a critical role in the development of the hypothalamus, pituitary, adrenal, and gonads.
GnRH receptor mutations are also associated with hypogonadotropic hypogonadism. The GnRH receptor is a G protein–coupled receptor for GnRH. Patients with GnRH mutations have a spectrum of reproductive dysfunction from partial to complete hypogonadism.
Mutations in the PC1 or convertase-1 gene are associated with hypogonadotropic hypogonadism in conjunction with obesity and diabetes mellitus. PC1 is essential in the cleavage of multiple propeptides to their active peptide hormone. The gene is believed to have a role in GnRH secretion and release.
Prader-Willi syndrome is caused either by mutations or deletions of a specific locus within paternal chromosome 15 or, less commonly, by maternal uniparental disomy (two maternal copies) of this locus. Symptoms include obesity, mild or moderate mental retardation, infantile hypotonia, and hypogonadotropic hypogonadism.
Hemochromatosis is associated with treatable hypogonadotropic hypogonadism; some men with hemochromatosis develop primary testicular failure.
Biologically inactive LH or FSH can result from genetic mutations in either the hormones or their receptors. The mutations result in a spectrum of dysfunction from complete virilization failure to less severe hypogonadism.
Pituitary mass lesions are uncommon but are recognized causes of hypogonadotropic hypogonadism and male infertility. Such lesions interfere with the release of LH and FSH, either by direct compression of the portal system or by decreasing secretion of these gonadotropins.
In hyperprolactinemia, the elevated serum prolactin level causes hypogonadism because it interferes with the normal pulsatile release of GnRH. Adenomas of the pituitary can cause hyperprolactinemia (due to infundibular compression and resultant inhibition of hypothalamic dopamine that tonically inhibits prolactin synthesis and secretion), together with headaches and visual field impairment (due to direct compression on the optic chiasm). Selective serotonin reuptake inhibitors can also cause hyperprolactinemia.
Spermatogenesis is dependent on a high androgen concentration. Genetic steroidogenic enzyme deficiencies can result in combined defects in multiple steroid hormones including testosterone and/or DHT. Androgen deficiency results in a spectrum of phenotypic abnormalities ranging from incomplete virilization to completely feminized genitalia and cryptorchid testes. Alternatively, in congenital adrenal hyperplasia, impaired corticosteroid and androgenic steroid synthesis often results in ACTH-dependent elevations in adrenal androgens (see Chapter 21).
The X-linked androgen receptor (AR) is a nuclear steroid receptor that is classically activated upon androgen binding to facilitate transcriptional activation of a host of target genes. Androgen insensitivity syndromes result from mutations in AR structure and/or function. Complete loss of AR function results in complete feminization of 46,XY individuals. Because testosterone is converted to estradiol by peripheral aromatization, estradiol levels are usually elevated and feminization occurs in a similar fashion to normal XX females at the time of puberty. In less severe cases, the phenotypic spectrum ranges from simple male infertility to ambiguous genitalia and hypospadias.
Anabolic steroid abuse results in negative feedback at the level of the hypothalamus and pituitary, and LH and FSH release is reduced. This in turn disables endogenous testosterone production and spermatogenesis because normal spermatogenesis requires both FSH and adequate intratesticular testosterone. Decreased testicular size and gynecomastia can also be seen in association with long-time anabolic steroid abuse. The extent and reversibility of these detrimental effects depend on dose and duration of use. Normal hormonal function usually returns after these agents are discontinued.
A number of conditions damage spermatogenic potential by direct effects on the testicles.
Varicoceles are considered the most common cause of subfertility in men. The term varicocele refers to abnormally dilated scrotal veins. A varicocele is present in about 15% of the normal male population, but in approximately 40% of men presenting with infertility.
Possible pathogenic mechanisms in varicocele formation include the anatomical configuration of the left internal spermatic vein, incompetent or absent valves, and potential for a partial left renal vein compression between the aorta and the superior mesenteric artery. An acute varicocele can also be caused by retroperitoneal malignancies with arteriovenous shunting into the venous system.
Varicoceles are associated with impaired spermatogenesis by one of several mechanisms: increased scrotal temperatures, alterations in testicular blood flow, reduced testicular size, overproduction of adrenal steroid metabolites, increased oxidative stress, which may damage cell membrane integrity or cause DNA damage, and alterations in the hypothalamic-pituitary-gonadal axis, leading to decreased serum testosterone levels. The pathophysiology of the impaired spermatogenesis is likely multifactorial in many cases.
Several studies have shown decreased semen quality and increased sperm DNA damage in varicocele patients compared with normal controls. However, the evidence for a clinical benefit of varicocele repair in improving fertility is controversial.
Genetic disorders are characterized as (a) abnormalities of entire chromosomes (abnormalities of the karyotype), (b) deletions of specific areas of chromosomes, or (c) specific mutations within genes. These disorders can alter spermatogenesis and impair normal development of the genital tract, thus decreasing fertilization capacity.
Chromosomal defects are categorized as either numerical or structural. Numerical chromosome abnormalities include deletion or duplication of whole chromosomes. Structural chromosome abnormalities include deletion, inversion, or duplication of a portion of a chromosome, or translocation of part of one chromosome to another chromosome. Both autosomal and sex chromosomes may be affected. Such abnormalities occur with much greater frequency in infertile men than in the general population. About 1 in 20 infertile men has a chromosomal abnormality, and most of these cases involve a sex chromosome. These men are usually azoospermic or severely oligospermic.
Klinefelter syndrome (47,XXY) is the most common chromosomal disorder associated with infertility. Patients with Klinefelter syndrome are severely oligospermic or azoospermic. The phenotype of men with Klinefelter syndrome varies but can include increased height, female hair distribution, gynecomastia, decreased level of intelligence, diabetes mellitus, obesity, increased incidence of leukemia and nonseminomatous extragonadal germ cell tumors, small firm testes, and infertility. Laboratory studies show increased serum FSH, normal or increased serum estradiol, and normal or low serum testosterone (with a tendency to decrease with age). Leydig cell function is commonly impaired in men with Klinefelter syndrome. Patients with Klinefelter syndrome who are mosaics with 46,XY/47,XXY have a less severe phenotype, with a variable sperm production.
There are other less common chromosomal defects. Most patients with mixed gonadal dysgenesis have a mosaic karyotype of 45,X/46,XY, but others have a normal 46,XY. Affected individuals can have male, female, or ambiguous genitalia, streak gonads, or normal testes. The XX male syndrome (46,XX) is caused by translocation of the SRY sex-determination gene from the paternal Y chromosome to the paternal X of the offspring, resulting in “normal” development of testes in the XX fetus, but lack of all spermatogenic genes normally found on the Y chromosome. The XYY male syndrome (47,XYY) is characterized by decreased intelligence, antisocial behavior, an increased incidence of leukemia, and impairment of spermatogenesis.
Microdeletions of the Y chromosome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


