Figure 11-1. Arousal spectrum of sleep and wakefulness. One’s state of arousal is more complicated than simply being “awake” or “asleep.” Rather, arousal exists as if on a dimmer switch, with many phases along the spectrum. Where on the spectrum one lies is influenced in large part by five key neurotransmitters: histamine (HA), dopamine (DA), norepinephrine (NE), serotonin (5HT), and acetylcholine (ACh). When there is good balance between too much and too little arousal (depicted by the gray [baseline] color of the brain), one is awake, alert, and able to function well. As the dial shifts to the right there is too much arousal, which may cause hypervigilance and consequently insomnia at night. As arousal further increases this can cause cognitive dysfunction, panic, and in extreme cases perhaps even hallucinations. On the other hand, as arousal diminishes, individuals may experience inattentiveness, cognitive dysfunction, sleepiness, and ultimately sleep.
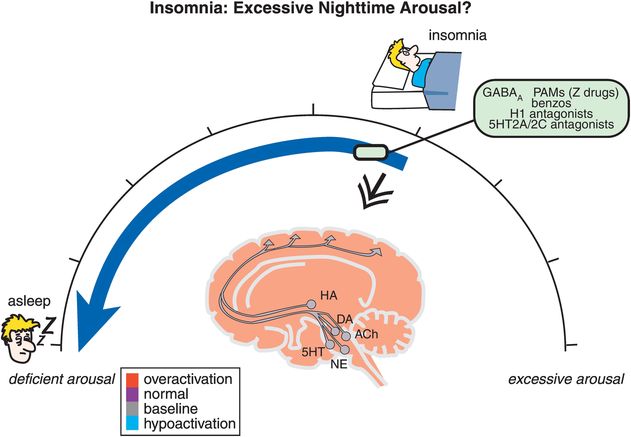
Figure 11-2. Insomnia: excessive nighttime arousal? Insomnia is conceptualized as being related to hyperarousal at night, depicted here as the brain being red (overactive). Agents that reduce brain activation, such as positive allosteric modulators of GABAA receptors (e.g., benzodiazepines, “Z drugs”), histamine 1 antagonists, and serotonin 2A/2C antagonists, can shift one’s arousal state from hyperactive to sleep.
On the other hand, as arousal diminishes, symptoms crescendo from mere inattentiveness to more severe forms of cognitive disturbances until the patient has excessive daytime sleepiness with sleep attacks (Figure 11-1, and hypoactivation of the brain in blue at the left-hand side of the spectrum in Figure 11-3). From a treatment perspective, sleepiness can be conceptualized as a disorder of deficient daytime arousal, with wake-promoting agents moving the patient from too little arousal to awake with normal alertness (Figure 11-3).
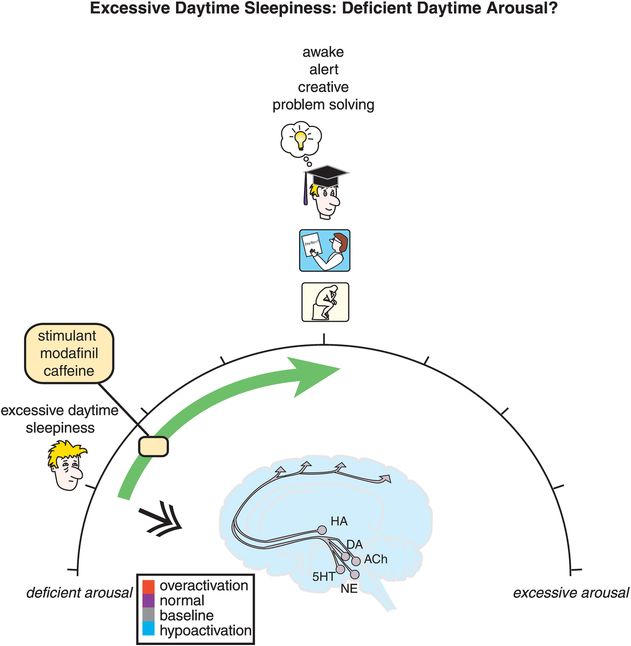
Figure 11-3. Excessive daytime sleepiness: deficient daytime arousal. Excessive sleepiness is conceptualized as being related to hypoarousal during the day, depicted here as the brain being blue (hypoactive). Agents that increase brain activation, such as the stimulants, modafinil, and caffeine, can shift one’s arousal state from hypoactive to awake with normal alertness.
Note in Figure 11-1 that cognitive disturbance is the product of both too little and too much arousal, consistent with the need of cortical pyramidal neurons to be optimally “tuned,” with too much activity making them just as out of tune as too little. Note also in Figures 11-1 through 11-3 that the arousal spectrum is linked to the actions of five neurotransmitters shown in the brains represented in these figures (i.e., histamine, dopamine, norepinephrine, serotonin, and acetylcholine). Sometimes these neurotransmitter circuits as a group are called the ascending reticular activating system, because they are known to work together to regulate arousal. This same ascending neurotransmitter system is blocked at several sites by many agents that cause sedation. Actions of sedating drugs on these neurotransmitters are discussed in Chapter 5 on antipsychotics and illustrated in Figure 5-38. Figure 11-1 also shows that excessive arousal can extend past insomnia to panic, hallucinations, and all the way to frank psychosis (far right-hand side of the spectrum).
The sleep/wake switch
We have discussed how the ascending neurotransmitter systems from the brainstem regulate a cortical arousal system on a smooth continuum like a rheostat on a lighting system or a volume button on a radio. There is another set of circuits in the hypothalamus that regulate sleep and wake discontinuously, like an on/off switch. Not surprisingly, this circuitry is called the sleep/wake switch (Figure 11-4). The “on” switch is known as the wake promoter and is localized within the tuberomammillary nucleus (TMN) of the hypothalamus (Figure 11-4A). The “off” switch is known as the sleep promoter and is localized within the ventrolateral preoptic (VLPO) nucleus of the hypothalamus (Figure 11-4B).
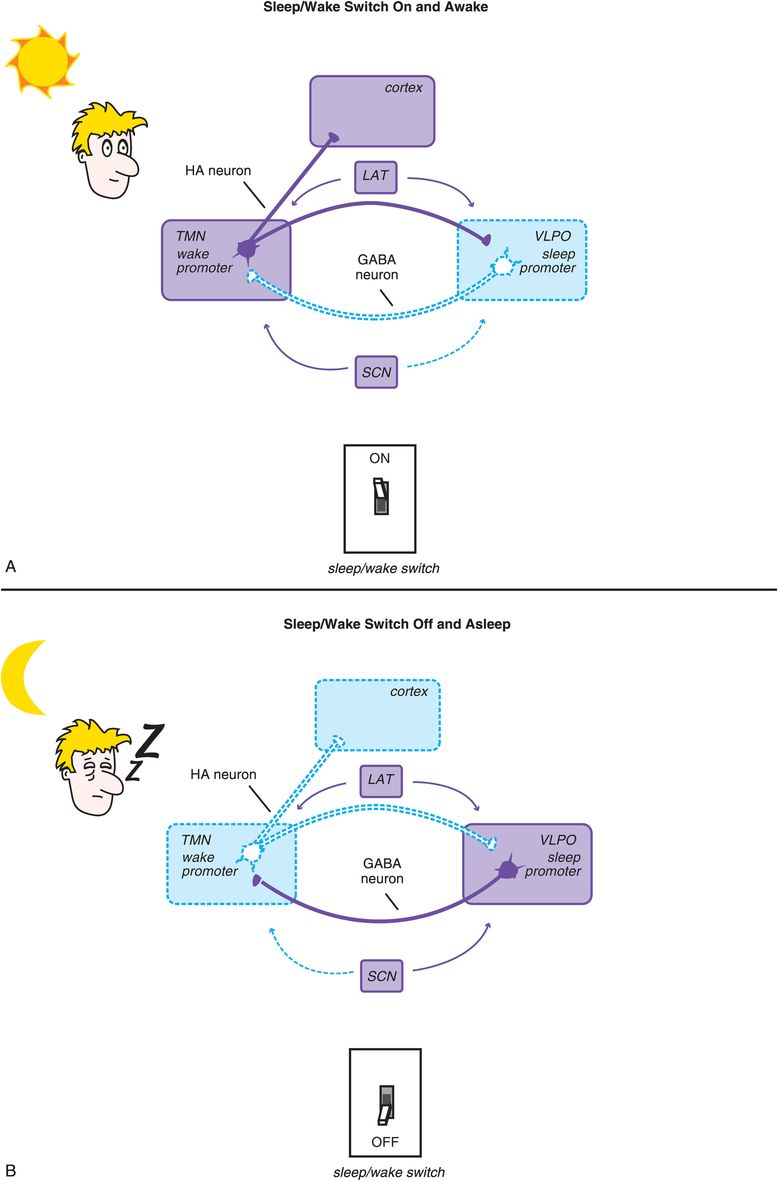
Figure 11-4. Sleep/wake switch. The hypothalamus is a key control center for sleep and wake, and the specific circuitry that regulates sleep/wake (i.e., whether the dimmer switch is set all the way to the left for sleep or is somewhere else along the continuum for wake) is called the sleep/wake switch. The “off” setting, or sleep promoter, is localized within the ventrolateral preoptic nucleus (VLPO) of the hypothalamus, while “on” – the wake promoter – is localized within the tuberomammillary nucleus (TMN) of the hypothalamus. Two key neurotransmitters regulate the sleep/wake switch: histamine from the TMN and GABA from the VLPO. (A) When the TMN is active and histamine is released to the cortex and the VLPO, the wake promoter is on and the sleep promoter inhibited. (B) When the VLPO is active and GABA is released to the TMN, the sleep promoter is on and the wake promoter inhibited. The sleep/wake switch is also regulated by orexin/hypocretin neurons in the lateral hypothalamus (LAT), which stabilize wakefulness, and by the suprachiasmatic nucleus (SCN) of the hypothalamus, which is the body’s internal clock and is activated by melatonin, light, and activity to promote either sleep or wake.
Two other sets of neurons are shown in Figure 11-4 as regulators of the sleep/wake switch: orexin-containing neurons of the lateral hypothalamus (LAT) and melatonin-sensitive neurons of the suprachiasmatic nucleus (SCN). The lateral hypothalamus serves to stabilize and promote wakefulness via a peptide neurotransmitter known by two different names: orexin and hypocretin. These lateral hypothalamic neurons and their orexin are lost in narcolepsy, especially narcolepsy with cataplexy. New hypnotics on the horizon (orexin antagonists) block the receptors for these neurotransmitters and are discussed later in this chapter. The SCN is the brain’s internal clock, or pacemaker, and regulates circadian input to the sleep/wake switch in response to how it is programmed by hormones such as melatonin and by the light/dark cycle. Circadian rhythms and the SCN are discussed in Chapter 7 on antidepressants and illustrated in Figures 7-39 to 7-42.
The circadian wake drive is shown in Figure 11-5 over two full 24-hour cycles. Also shown in Figure 11-5 is the ultradian sleep cycle (a cycle faster than a day, showing cycling in and out of REM and slow-wave sleep several times during the night). Homeostatic sleep drive, illustrated as well in Figure 11-5, increases the drive for sleep as the day goes on, presumably due to fatigue, and diminishes at night with rest. The novel neurotransmitter adenosine is linked to homeostatic drive, and appears to accumulate as this drive increases during the day, and to diminish at night. Caffeine is now known to be an antagonist of adenosine, and this may explain in part its ability to promote wakefulness and diminish fatigue, namely by opposing endogenous adenosine’s regulation of the homeostatic sleep drive.
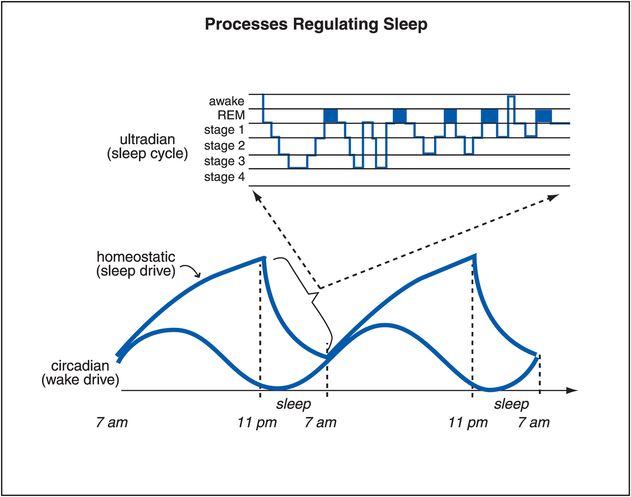
Figure 11-5. Processes regulating sleep. Several processes that regulate sleep/wake are shown here. The circadian wake drive is a result of input (light, melatonin, activity) to the suprachiasmatic nucleus. Homeostatic sleep drive increases the longer one is awake and decreases with sleep. As the day progresses, circadian wake drive diminishes and homeostatic sleep drive increases until a tipping point is reached and the ventrolateral preoptic sleep promoter (VLPO) is triggered to release GABA in the tuberomammillary nucleus (TMN) and inhibit wakefulness. Sleep itself consists of multiple phases that recur in a cyclical manner; this process is known as the ultradian cycle, and is depicted at the top of this figure.
Two key neurotransmitters regulate the sleep/wake switch: histamine from the TMN and GABA (γ-aminobutyric acid) from the VLPO. Thus, when the sleep/wake switch is on, the wake promoter TMS is active and histamine is released (Figure 11-4). This occurs both in the cortex to facilitate arousal, and in the VLPO to inhibit the sleep promoter. As the day progresses, circadian wake drive diminishes and homeostatic sleep drive increases (Figure 11-5); eventually a tipping point is reached, and the VLPO sleep promoter is triggered, the sleep/wake switch is turned off, and GABA is released in the TMN to inhibit wakefulness (Figure 11-4).
Disorders characterized by excessive daytime sleepiness can be conceptualized as the sleep/wake switch being off during the daytime. Wake-promoting treatments such as modafinil given during the day tip the balance back to wakefulness by promoting the release of histamine from TMN neurons. The exact mechanism of this enhancement of histamine release by modafinil or stimulants is not known, but is currently hypothesized to be related in part to a downstream consequence of the actions of wake-promoting drugs on dopamine neurons, especially by blocking the dopamine transporter DAT.
On the other hand, disorders characterized by insomnia can be conceptualized as the sleep/wake switch being on at night. Insomnia can be treated either by agents that enhance GABA actions, and thus inhibit the wake promoter, or by agents that block the action of histamine released from the wake promoter and act at postsynaptic H1 receptors.
Disorders characterized by a disturbance in circadian rhythm can be conceptualized as either “phase delayed,” with the wake promoter and sleep/wake switch being turned on too late in a normal 24-hour cycle, or “phase advanced,” with the wake promoter and sleep/wake switch being turned on too early in a normal 24-hour cycle. That is, individuals who are phase delayed, including many depressed patients and many normal adolescents, still have their sleep/wake switch off when it is time to get up (see discussion in Chapter 7 and Figure 7-39). Giving such individuals morning light and evening melatonin can reset the circadian clock in the SCN so that it wakes the person up earlier. Other individuals may be phase advanced, including many normal elderly people. Giving these individuals evening light and morning melatonin can reset their SCNs so that the sleep/wake switch stays off a bit longer, returning the patient to a normal rhythm.
Histamine
Histamine is one of the key neurotransmitters regulating wakefulness, and is the ultimate target of many wake-promoting drugs (via downstream histamine release) and sleep-promoting drugs (antihistamines). Histamine is produced from the amino acid histidine, which is taken up into histamine neurons and converted to histamine by the enzyme histidine decarboxylase (Figure 11-6). Histamine’s action is terminated by two enzymes working in sequence: histamine N-methyl-transferase, which converts histamine to N-methyl-histamine, and MAO-B, which converts N-methyl-histamine into N-MIAA (N-methyl-indole-acetic acid), an inactive substance (Figure 11-7). Additional enzymes such as diamine oxidase can also terminate histamine action outside of the brain. Note that there is no apparent reuptake pump for histamine. Thus, histamine is likely to diffuse widely away from its synapse, just like dopamine does in prefrontal cortex.
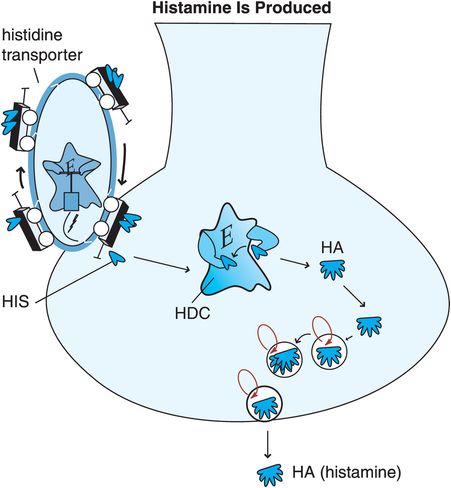
Figure 11-6. Histamine is produced. Histidine (HIS), a precursor to histamine, is taken up into histamine nerve terminals via a histidine transporter and converted into histamine by the enzyme histidine decarboxylase (HDC). After synthesis, histamine is packaged into synaptic vesicles and stored until its release into the synapse during neurotransmission.
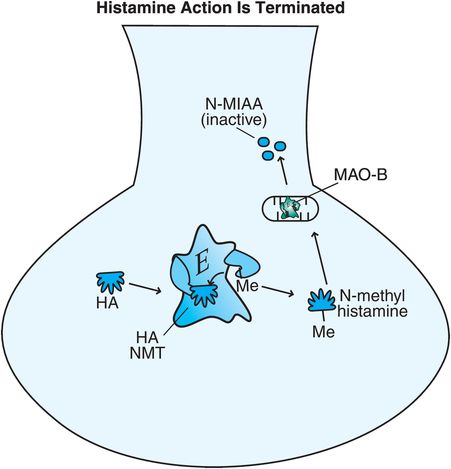
Figure 11-7. Histamine’s action is terminated. Histamine can be broken down intracellularly by two enzymes. Histamine N-methyl-transferase (histamine NMT) converts histamine into N-methyl-histamine, which is then converted by monoamine oxidase B (MAO-B) into the inactive substance N-methyl-indole-acetic acid (N-MIAA).
There are a number of histamine receptors (Figures 11-8 through 11-11). The postsynaptic histamine 1 (H1) receptor is best known (Figure 11-9A) because it is the target of “antihistamines” (i.e., H1 antagonists) (Figure 11-9B). When histamine itself acts at H1 receptors, it activates a G-protein-linked second-messenger system that activates phosphatidyl inositol, and the transcription factor cFOS, and results in wakefulness, normal alertness, and pro-cognitive actions (Figure 11-9A). When these H1 receptors are blocked in the brain, they interfere with the wake-promoting actions of histamine, and thus can cause sedation, drowsiness, or sleep (Figure 11-9B).
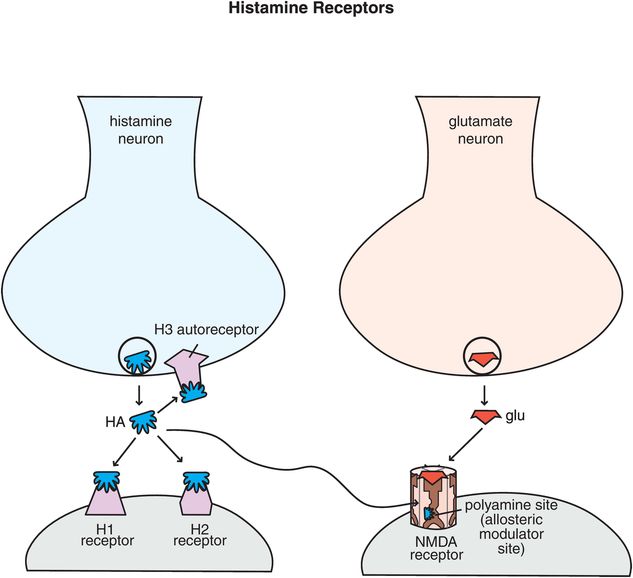
Figure 11-8. Histamine receptors. Shown here are receptors for histamine that regulate its neurotransmission. Histamine 1 and histamine 2 receptors are postsynaptic, while histamine 3 receptors are presynaptic autoreceptors. There is also a binding site for histamine on NMDA receptors – it can act at the polyamine site, which is an allosteric modulatory site.
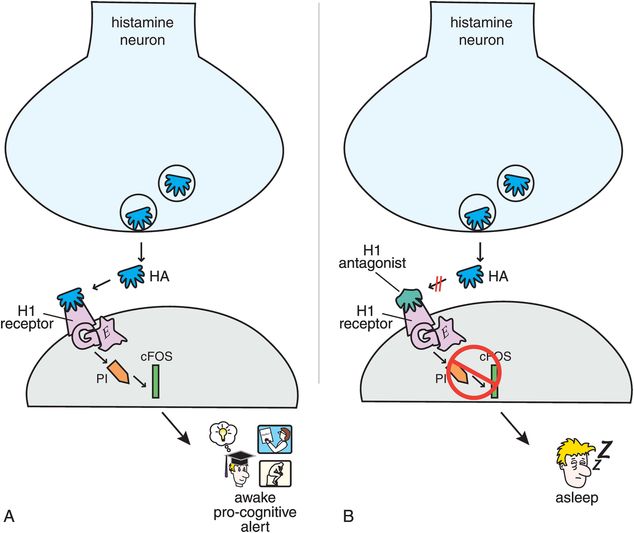
Figure 11-9. Histamine 1 receptors. (A) When histamine binds to postsynaptic histamine 1 receptors, it activates a G-protein-linked second-messenger system that activates phosphatidyl inositol and the transcription factor cFOS. This results in wakefulness and normal alertness. (B) Histamine 1 antagonists prevent activation of this second messenger and thus can cause sleepiness.
Histamine 2 (H2) receptors, best known for their actions in gastric acid secretion and the target of a number of anti-ulcer drugs, also exist in the brain (Figure 11-10). These postsynaptic receptors also activate a G-protein second-messenger system with cAMP, phosphokinase A, and the gene product CREB. The function of H2 receptors in brain is still being clarified, but apparently is not linked directly to wakefulness.
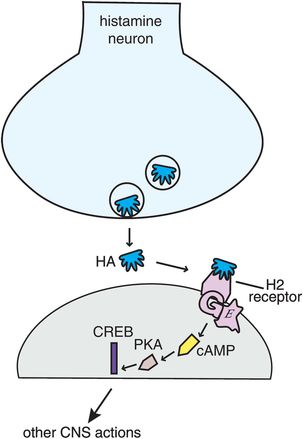
Figure 11-10. Histamine 2 receptors. Histamine 2 receptors are present both in the body and in the brain. When histamine binds to postsynaptic histamine 2 receptors it activates a G-protein-linked second-messenger system with cAMP, phosphokinase A, and the gene product CREB. The function of histamine 2 receptors in the brain is not yet elucidated but does not appear to be directly linked to wakefulness.
A third histamine receptor is present in brain, namely the H3 receptor (Figures 11-8 and 11-11). Synaptic H3 receptors are presynaptic (Figure 11-11A) and function as autoreceptors (Figure 11-11B). That is, when histamine binds to these receptors, it turns off further release of histamine (Figure 11-11B). One novel approach to new wake-promoting and pro-cognitive drugs is to block these receptors, thus facilitating the release of histamine, allowing histamine to act at H1 receptors to produce the desired effects (Figure 11-11C). Several H3 antagonists are in clinical development.
There is a fourth type of histamine receptor, H4, but these are not known to occur in the brain. Finally, histamine acts also at NMDA (N-methyl-D-aspartate) receptors (Figure 11-8). Interestingly, when histamine diffuses away from its synapse to a glutamate synapse containing NMDA receptors, it can act at an allosteric modulatory site called the polyamine site, to alter the actions of glutamate at NMDA receptors (Figure 11-8). The role of histamine and function of this action are not well clarified.
Histamine neurons all arise from a single small area of the hypothalamus known as the tuberomammillary nucleus (TMN), which is part of the sleep/wake switch illustrated in Figure 11-4. Thus, histamine plays an important role in arousal, wakefulness, and sleep. The TMN is a small bilateral nucleus that provides histaminergic input to most brain regions and to the spinal cord (Figure 11-12).
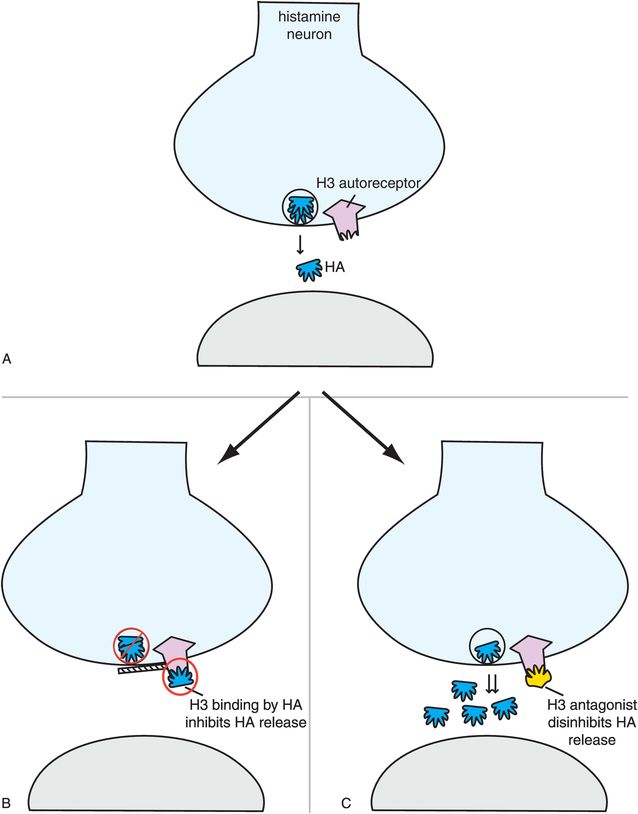
Figure 11-11. Histamine 3 receptors. Histamine 3 receptors are presynaptic autoreceptors (A), which means that when histamine binds to these receptors it turns off further histamine release (B). Antagonists of these receptors, which are in development, therefore disinhibit histamine release (C) and may hypothetically enhance alertness and cognition.
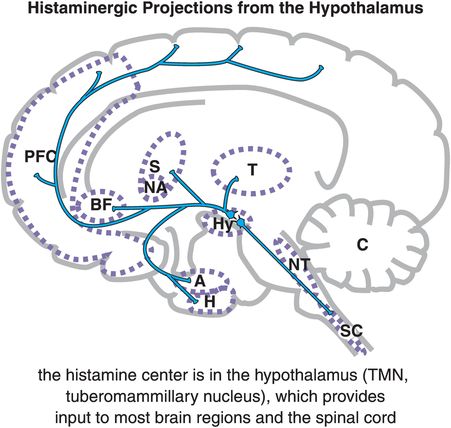
Figure 11-12. Histaminergic projections from the hypothalamus. In the brain, histamine (HA) is produced solely by cells in the tuberomammillary nucleus (TMN) of the hypothalamus (Hy). From the TMN, histaminergic neurons project to various brain regions including the prefrontal cortex (PFC), the basal forebrain (BF), the striatum (S) and nucleus accumbens (NA), the amygdala (A) and hippocampus (H), brainstem neurotransmitter centers (NT), and spinal cord (SC).
Insomnia and hypnotics
What is insomnia?
Insomnia has many causes, including both sleep disorders and psychiatric disorders. Insomnia can also contribute to the onset, exacerbation, or relapse of many psychiatric disorders and is linked to various dysfunctions in many medical illnesses. Primary insomnia may be a condition with too much arousal both at night and during the day, and thus may be a form of insomnia where the patient is not sleepy during the day despite having poor sleep at night. Primary insomnia may also be a symptom that can progress to a first major depressive episode. Thus, is insomnia a symptom or a disorder? The answer appears to be “yes, both.”
Chronic treatment for chronic insomnia?
A major reconceptualization of insomnia has recently occurred among experts, with a newly formed consensus that insomnia can be chronic and that it may need to be treated chronically. This is a departure from the position held by many sleep experts in the past – that insomnia was treated by attacking its underlying cause, and not by giving chronic “symptomatically masking” treatment with hypnotics. The old guidelines recommending short-term use of hypnotics for insomnia were the product of safety concerns for hypnotics identified first during the barbiturate era and then during the benzodiazepine era.
Other problems associated with long-term use of hypnotics have to do with use of drugs whose half-lives are not ideal for use as hypnotics (Figure 11-13A, B, and C). That is, many agents used as hypnotics, particularly in the past, have half-lives that are too long (Figure 11-13A and B). This can cause drug accumulation and hip fractures from falls, especially in the elderly, when such agents are used every night (Figure 11-13A). Long half-life can also cause next-day carryover effects and sedation and memory problems from residual daytime drug levels (Figure 11-13A and B). Other agents used as hypnotics have half-lives that are too short, and their effects can wear off before it is time to wake up, causing insufficient sleep maintenance and nocturnal awakenings, as well as restless and disturbed sleep in some patients (Figure 11-13C). More recently, however, the hypnotics given most frequently for chronic use are those that have optimized half-lives targeting rapid onset of action, and plasma drug levels above the minimally effective concentration, but only until it is time to wake up (Figure 11-13D). Perhaps no therapeutic area of psychopharmacology is as critically dependent upon plasma drug levels, and thus the pharmacokinetics of the drug, as is the use of hypnotics. This fact may be related to the nature of the arousal system and of the sleep/wake switch, which requires pharmacologic action to a degree sufficient to reach the critical tipping point that trips the switch “off” to allow sleep, but only at night.
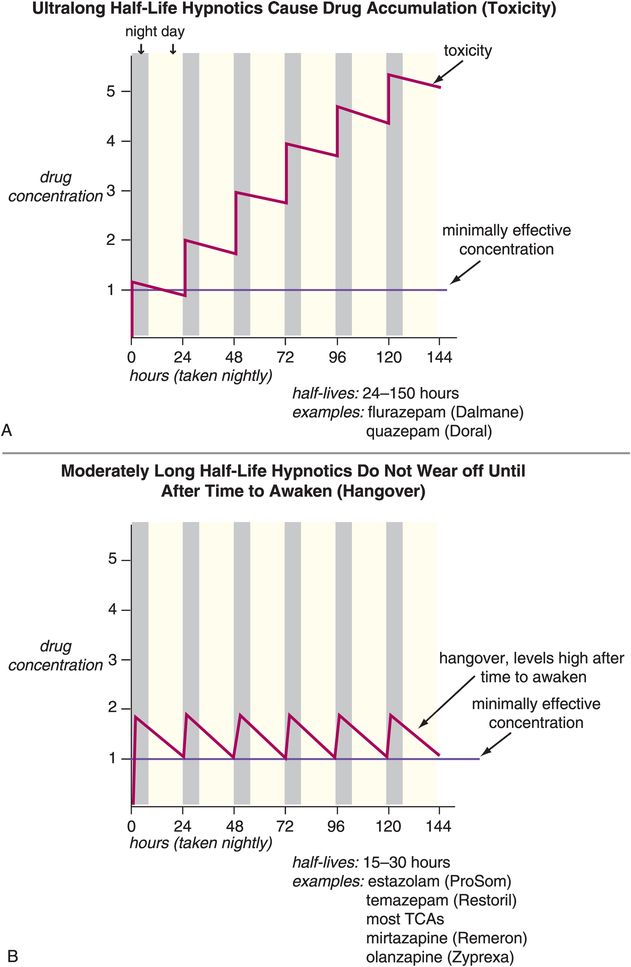
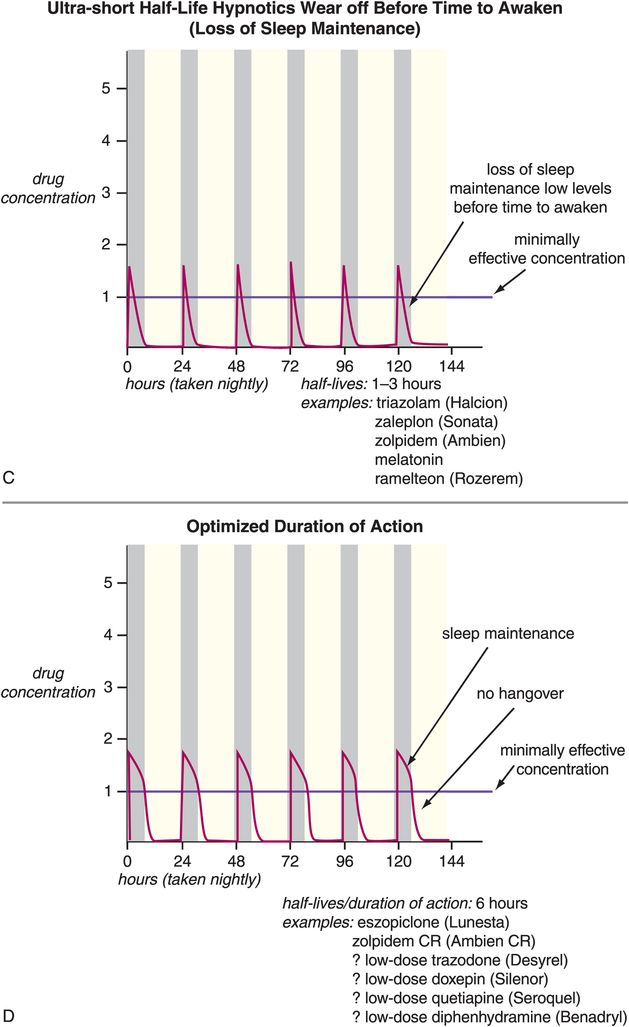
Figure 11-13. Half-lives of hypnotics. The half-lives of hypnotics can have an important impact on their tolerability and efficacy profiles. (A) Hypnotics with ultra-long half-lives (greater than 24 hours: for example, flurazepam and quazepam) can cause drug accumulation with chronic use. This can cause impairment that has been associated with increased risk of falls, particularly in the elderly. (B) Hypnotics with moderate half-lives (15–30 hours: estazolam, temazepam, most tricyclic antidepressants, mirtazapine, olanzapine) may not wear off until after the individual needs to awaken and thus may have “hangover” effects (sedation, memory problems). (C) Hypnotics with ultra-short half-lives (1–3 hours: triazolam, zaleplon, zolpidem, melatonin, ramelteon) can wear off before the individual needs to awaken and thus cause loss of sleep maintenance. (D) Hypnotics with half-lives that are short but not ultra-short (approximately 6 hours: zolpidem CR and perhaps low doses of trazodone or doxepin) may provide rapid onset of action and plasma levels above the minimally effective concentration only for the duration of a normal night’s sleep.
Other reasons for short-term restrictions on benzodiazepine hypnotics (Figure 11-14) in the past had to do with their long-term effects, including loss of efficacy over time (tolerance) and withdrawal effects, including rebound insomnia in some patients worse than their original insomnia (Figure 11-15A). Recent investigations have shown that some non-benzodiazepine hypnotics may not have these problems (Figure 11-15B). These include the GABAA positive allosteric modulators (PAMs), sometimes also called “Z drugs” (because they all start with the letter Z: zaleplon, zolpidem, zopiclone) (Figure 11-16). Perhaps the best long-term studies have been done with eszopiclone, which shows little or no tolerance, dependence, or withdrawal with use for many months (Figure 11-15B). This is probably also the case for long-term use of zolpidem, zolpidem CR, and the melatonergic agent ramelteon, as well as for “off-label” use of the sedating antidepressant trazodone, none of which have restrictions against chronic use. For these reasons, it is now recognized that chronic insomnia may need chronic treatment with certain hypnotics.
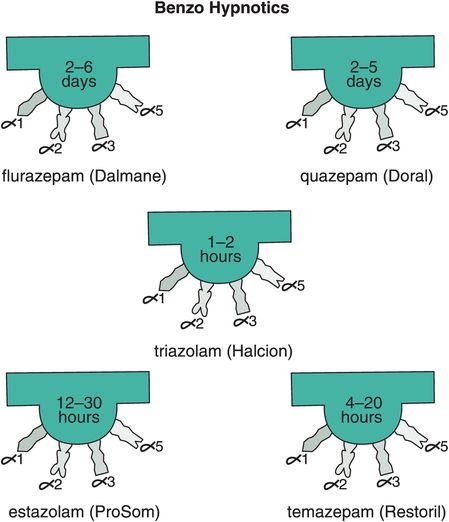
Figure 11-14. Benzo hypnotics. Five benzodiazepines that are approved in the United States for insomnia are shown here. These include flurazepam and quazepam, which have ultra-long half-lives; triazolam, which has an ultra-short half-life; and estazolam and temazepam, which have moderate half-lives.
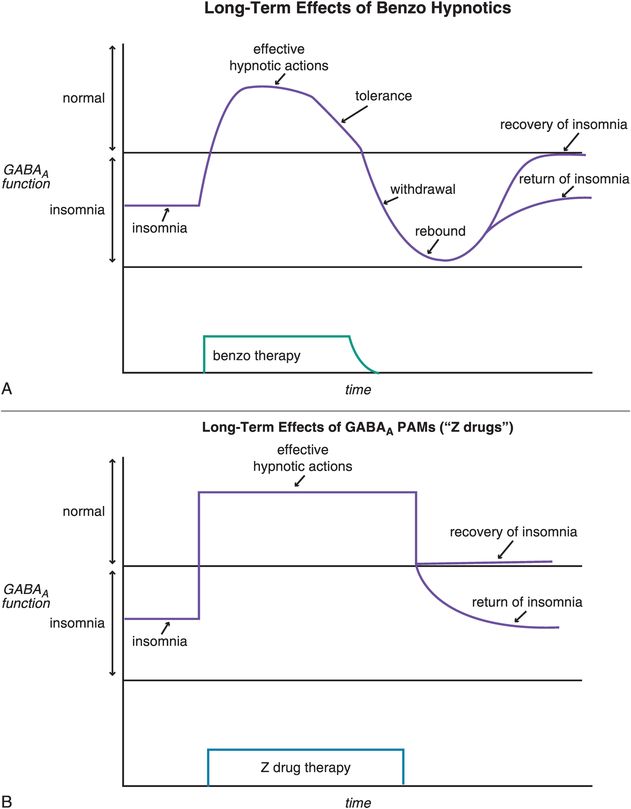
Figure 11-15. Long-term effects of hypnotics. (A) Short-term, benzodiazepines can be efficacious for treating insomnia. With long-term use, however, benzodiazepines may cause tolerance and, if discontinued, withdrawal effects that may include rebound insomnia. (B) Positive allosteric modulators (PAMs) at GABAA receptors are efficacious for insomnia in the short term, and in the long term do not seem to cause tolerance or withdrawal effects.
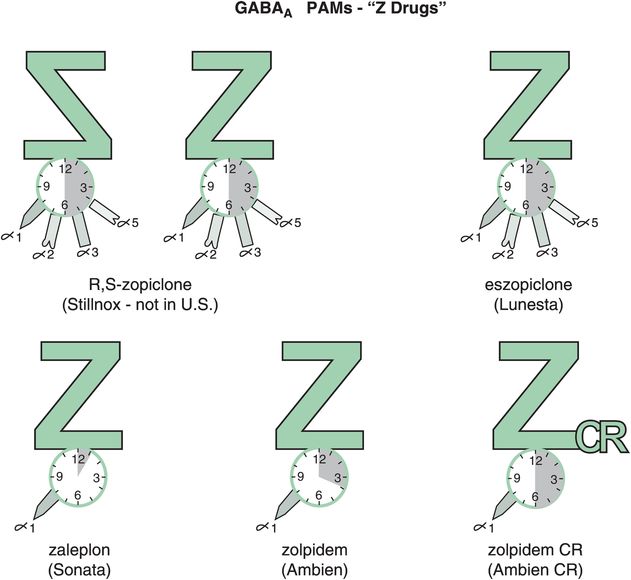
Figure 11-16. GABAA positive allosteric modulators (PAMs). Several GABAA PAMs, or “Z drugs,” are shown here. These include racemic zopiclone (not available in the United States), eszopiclone, zaleplon, zolpidem, and zolpidem CR. Zaleplon, zolpidem, and zolpidem CR are selective for GABAA receptors that contain the α1 subunit; however, it does not appear that zopiclone or eszopiclone have this same selectivity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


