Disorders of Calcium and Phosphorus Homeostasis
KEY CONCEPTS
![]() Severe acute hypercalcemia can result in cardiac arrhythmias, whereas chronic hypercalcemia can lead to calcium deposition in soft tissues including blood vessels and the kidney.
Severe acute hypercalcemia can result in cardiac arrhythmias, whereas chronic hypercalcemia can lead to calcium deposition in soft tissues including blood vessels and the kidney.
![]() The correction of hypercalcemia can include multiple pharmacotherapeutic modalities such as hydration, diuretics, bisphosphonates, and steroids, depending on the etiology and acuity of the hypercalcemia.
The correction of hypercalcemia can include multiple pharmacotherapeutic modalities such as hydration, diuretics, bisphosphonates, and steroids, depending on the etiology and acuity of the hypercalcemia.
![]() Hypocalcemia is typically associated with an insidious onset; however, some drugs such as cinacalcet are associated with rapid decreases in serum calcium.
Hypocalcemia is typically associated with an insidious onset; however, some drugs such as cinacalcet are associated with rapid decreases in serum calcium.
![]() Acute treatment of hypocalcemia requires calcium supplementation whereas chronic management may require other therapies such as vitamin D to maintain serum calcium values.
Acute treatment of hypocalcemia requires calcium supplementation whereas chronic management may require other therapies such as vitamin D to maintain serum calcium values.
![]() Hyperphosphatemia occurs most frequently in patients with chronic kidney disease (CKD).
Hyperphosphatemia occurs most frequently in patients with chronic kidney disease (CKD).
![]() Treatment of nonemergent hyperphosphatemia includes the use of phosphate binders to decrease absorption of phosphorus from the GI tract.
Treatment of nonemergent hyperphosphatemia includes the use of phosphate binders to decrease absorption of phosphorus from the GI tract.
![]() Hypophosphatemia is a relatively common complication among critically ill patients.
Hypophosphatemia is a relatively common complication among critically ill patients.
![]() Treatment of acute hypophosphatemia usually requires IV supplementation of phosphorous salts.
Treatment of acute hypophosphatemia usually requires IV supplementation of phosphorous salts.
INTRODUCTION
Disorders of calcium and phosphorus are common complications of multiple acute and chronic diseases. These disorders are frequently seen in the acute care setting; however, they are also often present in ambulatory patients, usually in a less severe state. The consequences of electrolyte disorders can range from asymptomatic to life-threatening, requiring hospitalization and emergent treatment. The maintenance of fluid and electrolyte homeostasis requires adequate functioning and modulation by multiple hormones on tissues of multiple organ systems.
There are many common drug therapies that can disturb the normal homeostatic mechanisms that maintain calcium and phosphorous balance. In addition, with some drug therapies, toxicity is enhanced when underlying electrolyte disorders are present. Drug-induced disorders typically respond well to discontinuation of the offending agent(s); however, additional therapies are sometimes required to correct the disorder. This chapter reviews the etiology, classification, clinical presentation, and therapy for the most common disorders of calcium and phosphorus homeostasis.
DISORDERS OF CALCIUM HOMEOSTASIS
The maintenance of physiologic calcium concentrations in the intracellular and extracellular spaces is vital for the preservation and function of cell membranes; propagation of neuromuscular activity; regulation of endocrine and exocrine secretory functions; blood coagulation cascade; platelet adhesion process; bone metabolism; muscle cell excitation/contraction coupling; and mediation of the electrophysiologic slow-channel response in cardiac and smooth-muscle tissue.
The disorders of calcium homeostasis are related to the calcium content of the extracellular fluid (ECF), which is tightly regulated and comprises less than 0.5% of the total body stores of calcium. Skeletal bone contains more than 99% of total body stores of calcium.1 ECF calcium is moderately bound to plasma proteins (46%), primarily albumin.2 Ionized or free calcium is the physiologically active form and is the fraction that is homeostatically regulated.3 Extracellular calcium, however, is most commonly measured as the total serum calcium level, which includes both bound and unbound calcium.2 The normal total calcium serum concentration range is 8.5 to 10.5 mg/dL (2.13 to 2.63 mmol/L).3
Proper assessment of total serum calcium concentrations includes measurement of the patient’s serum albumin concentration. Hypoalbuminemia, which can be associated with many chronic disease states, is probably the most common cause of “laboratory hypocalcemia.” Patients remain asymptomatic because the unbound or ionized fraction of serum calcium remains normal (normal range, 4.4 to 5.4 mg/dL [1.10 to 1.35 mmol/L]). A corrected total serum calcium (Sca) concentration can be calculated based on the measured total serum calcium and the difference between a patient’s measured albumin concentration and the normative value of 4 g/dL (40 g/L) by the following equations:
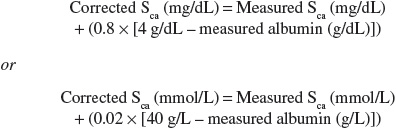
The concentration of ionized calcium is closely regulated by the interactions of parathyroid hormone (PTH), phosphorus, vitamin D, and calcitonin (Fig. 35-1). PTH increases serum calcium concentrations by stimulating calcium release from bone, increasing renal tubular reabsorption, and enhancing absorption in the GI tract secondary to increased renal production of 1,25-dihydroxy vitamin D3. Vitamin D directly increases serum calcium, as well as phosphorus concentrations, by increasing GI absorption. Indirectly, it can also lead to calcium release from bone and reduced renal excretion. Calcitonin inhibits osteoclastic bone resorption. Its plasma concentrations are increased when ionized calcium concentrations are high as the body attempts to return the calcium level to the normal range. Disruption of these homeostatic mechanisms results in the clinical manifestations of hypercalcemia or hypocalcemia.
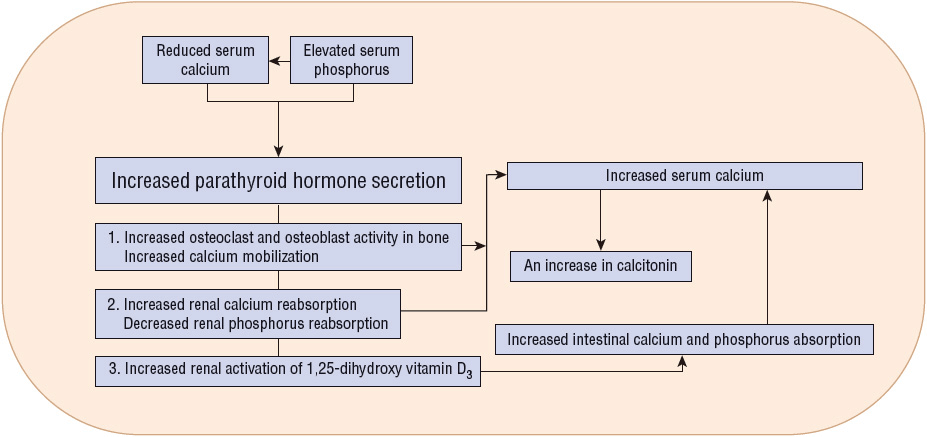
FIGURE 35-1 Homeostatic mechanisms to maintain serum calcium concentrations.
Alteration of the concentration of albumin or its binding of calcium can be expected to change the unbound fraction of total serum calcium. The most significant cause of changes in calcium binding to albumin is a change in ECF pH. In the presence of acute metabolic alkalosis the fraction of calcium bound to albumin is increased, thus reducing the plasma concentration of ionized calcium. This can result in symptomatic hypocalcemia; that is, paresthesia, muscle cramping and spasms, memory loss, and seizures.1 Conversely, metabolic acidosis decreases calcium binding to albumin and results in increased ionized calcium. Hypoalbuminemic states are probably the most common cause of “laboratory hypocalcemia.” When the albumin level is decreased, the ionized calcium concentration can be normal, although total serum calcium concentration is low. Each 1 g/dL (10 g/L) drop in the serum albumin concentration below 4 g/dL (40 g/L) will result in a decrease of total serum calcium concentration by 0.8 mg/dL (0.20 mmol/L).1,2 This approach of calculating an albumin-adjusted calcium concentration has been found to overestimate the degree of hypercalcemia and usually fails to identify hypocalcemia in critically ill patients; therefore, ionized calcium values should be used to assess calcium status in these patients.4,5
HYPERCALCEMIA
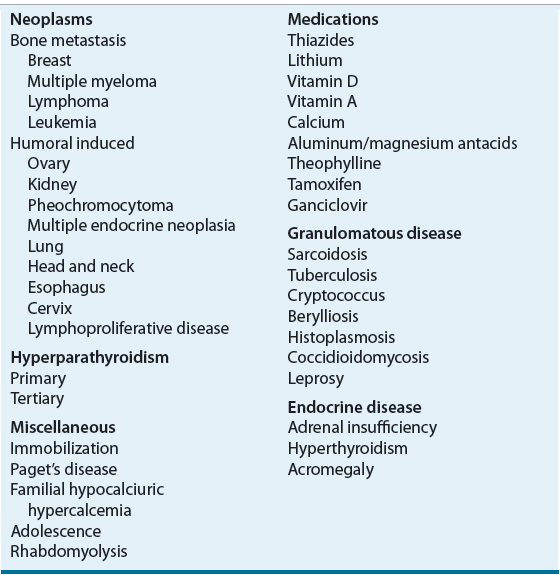
There are multiple and diverse causes of hypercalcemia (total serum calcium >10.5 mg/dL [>2.62 mmol/L]) (Table 35-1). The most common causes of hypercalcemia are cancer and primary hyperparathyroidism.
TABLE 35-1 Etiologies of Hypercalcemia
Epidemiology and Etiology
The reported incidence of primary hyperparathyroidism in the United States ranges from 10 to 30 cases per 100,000 people.6 Hypercalcemia of cancer occurs in approximately 20% to 40% of cancer patients at some time during the course of their disease.7 Cancer-associated hypercalcemia is predominantly encountered in hospitalized patients, whereas primary hyperparathyroidism accounts for the vast majority of cases in the outpatient setting.8,9
Pathophysiology
Hypercalcemia is the result of one or a combination of three primary mechanisms: increased bone resorption, increased GI absorption, or increased tubular reabsorption by the kidneys (see Fig. 35-1).
Many tumors secrete PTH-related protein (PTHrP), which binds to the PTH receptors in bone and renal tissues, leading to increased bone resorption and renal tubular reabsorption.11 Tumors can also secrete substances such as vitamin D, transforming growth factor, interleukins, prostaglandins, interferon, tumor necrosis factor, and granulocyte-macrophage colony-stimulating factor, which are associated with the development of hypercalcemia.7 Hypercalcemia of malignancy is a common complication of squamous cell carcinomas of the lung, head, and neck, hematologic malignancies such as multiple myeloma and T-cell lymphomas, and carcinomas of ovary, kidney, bladder, and breast. The most frequent types of malignancy associated with hypercalcemia are carcinomas of the lung and breast.7 Breast and squamous cell lung carcinomas secrete PTHrP which binds to the type I PTH receptor (PTHR1) and enhances bone resorption.10,11 In contrast, up to 40% of patients with multiple myeloma develop hypercalcemia principally as the result of osteoclast-mediated bone destruction.7
Primary hyperparathyroidism is the most common cause of chronic hypercalcemia in the general population. Benign parathyroid adenomas account for 80% to 85% of these cases of hyperparathyroidism, parathyroid hyperplasia accounts for 15%, and parathyroid carcinoma is the cause in less than 1% of cases.9
Other causes of chronic hypercalcemia include medications, endocrine and granulomatous disorders, physical immobilization, high bone-turnover states (adolescence and Paget’s disease), and rhabdomyolysis. Increased GI absorption can be the result of excessive ingestion of vitamin D analogs, calcium supplements, and lithium. Lithium and vitamin A therapy can increase bone resorption, whereas increased renal tubular reabsorption of calcium can occur with thiazide and lithium therapy. The exact mechanism of lithium-induced hypercalcemia is not known but may include competitive inhibition of calcium influx into cells, increasing the threshold sensitivity of the calcium-sensing receptor (CaSr) and subsequent inhibition of PTH gene transcription.8 Addison’s disease, acromegaly, and thyrotoxicosis are endocrine disorders that can lead to hypercalcemia because of increased renal tubular reabsorption and increased bone resorption. Finally, the granulomatous disorders (sarcoidosis, tuberculosis, histoplasmosis, and leprosy) are associated with hypercalcemia caused by an increase in GI and renal tubular absorption secondary to granuloma production of 1,25-dihydroxy vitamin D2.12 Milk-alkali syndrome is the term applied to those situations where an individual develops hypercalcemia following the ingestion of calcium and absorbable alkali (e.g., calcium carbonate) and is an important cause of hypercalcemia in patients who are not on dialysis.13,14
CLINICAL PRESENTATION Hypercalcemia
Clinical Presentation
Patients with mild-to-moderate hypercalcemia, that is, total serum calcium concentrations above the upper threshold of normal but less than 13 mg/dL (<3.25 mmol/L) or ionized calcium concentrations less than 6 mg/dL (<1.50 mmol/L) can often be asymptomatic. This is typically the case for the vast majority of patients who have drug-induced hypercalcemia or primary hyperparathyroidism.8,15,16 In fact, one study noted normocalcemia in approximately 20% of patients with a diagnosis of primary hyperparathyroidism, suggesting target tissue resistance to PTH.16
![]() The presenting signs and symptoms of severe hypercalcemia that occur if the total serum calcium concentration is >13 mg/dL (>3.25 mmol/L) may differ depending on the acuity of onset.2 Hypercalcemia of malignancy usually develops quickly and is accompanied by a classic symptom complex of anorexia, nausea and vomiting, constipation, polyuria, polydipsia, and nocturia.15 Polyuria and nocturia secondary to a urinary-concentrating defect constitute some of the most frequent renal effects of hypercalcemia.15 Hypercalcemic crisis is characterized by an acute elevation of total serum calcium to a value >15 mg/dL (>3.75 mmol/L), acute renal insufficiency, and obtundation (inability to arouse).15 If untreated, hypercalcemic crisis can progress to oliguric renal failure, coma, and life-threatening ventricular arrhythmias.15 The primary complications associated with chronic hypercalcemia (hyperparathyroidism) include metastatic calcification, hypercalciuria, and chronic renal insufficiency secondary to interstitial nephrocalcinosis.15
The presenting signs and symptoms of severe hypercalcemia that occur if the total serum calcium concentration is >13 mg/dL (>3.25 mmol/L) may differ depending on the acuity of onset.2 Hypercalcemia of malignancy usually develops quickly and is accompanied by a classic symptom complex of anorexia, nausea and vomiting, constipation, polyuria, polydipsia, and nocturia.15 Polyuria and nocturia secondary to a urinary-concentrating defect constitute some of the most frequent renal effects of hypercalcemia.15 Hypercalcemic crisis is characterized by an acute elevation of total serum calcium to a value >15 mg/dL (>3.75 mmol/L), acute renal insufficiency, and obtundation (inability to arouse).15 If untreated, hypercalcemic crisis can progress to oliguric renal failure, coma, and life-threatening ventricular arrhythmias.15 The primary complications associated with chronic hypercalcemia (hyperparathyroidism) include metastatic calcification, hypercalciuria, and chronic renal insufficiency secondary to interstitial nephrocalcinosis.15
Calcium and/or calcium–phosphorus complex deposition in blood vessels and multiple organs is a complication of chronic hypercalcemia and/or concomitant hyperphosphatemia and hyperparathyroidism. Calcium deposits in atherosclerotic lesions contribute to cardiac disease.17 Intracardiac and arterial calcifications have been found in patients with Paget’s disease who have normal renal function. It is hypothesized that similar calcification processes occur in both bone and vascular tissue, leading to cardiovascular diseases including heart failure, systolic hypertension, and ischemic heart disease.18
The electrocardiographic changes associated with hypercalcemia include shortening of the QT interval and coving of the ST-T wave.15 Very high serum calcium concentrations can cause T-wave widening, indicating a repolarization defect that may be associated with spontaneous ventricular tachyarrhythmias.15 Hypertension and arrhythmias have occurred in the setting of hypercalcemia. The effects of digoxin on cardiac conduction including lowering of the excitation threshold, shortening of the effective refractory period, and increased atrioventricular refractoriness can be potentiated by hypercalcemia.19
Nephrolithiasis (kidney stones) and nephrocalcinosis (calcium deposits in the kidney) are the primary renal complications arising from long-standing hypercalcemia, as the result of primary hyperparathyroidism. Stone formation is dependent on a favorable milieu within the kidney or urinary tract, such as oversaturation of the urine and/or reduced concentrations of endogenous inhibitors of crystal formation (e.g., citrate or pyrophosphate). It is estimated that hyperparathyroidism accounts for 2% to 8% of all patients with calcium stones.20,21 Of note, in those patients with low glomerular filtration rates (GFRs), the 24-hour urinary calcium will actually diminish secondary to decreased production of 1,25-dihydroxy vitamin D2. However, the fractional excretion of calcium might increase.21 Sarcoidosis is the other hypercalcemic condition frequently associated with calcium stones.20 Other causes of nephrolithiasis with calcium-containing stones include hypocitraturia, renal tubular acidosis, hyperoxaluria, and hyperuricosuria.22,23 Stone formers who have primary hyperparathyroidism are more likely to be female, older than 50 years of age, and have a family history of multiple endocrine disorders.20 High dietary sodium intake can also raise urinary calcium concentrations, perhaps due to a reduction in calcium reabsorption in the kidney, thus predisposing patients to calcium stones. Although chronic renal failure can be the ultimate result of persistent stones, it is the primary cause of renal disease in <2% of the end-stage renal disease population.
TREATMENT
Desired Outcome
The indications for the treatment of acute hypercalcemia are dependent on the severity of hypercalcemia, acuity of its development, and presence or absence of symptoms requiring emergent treatment (e.g., necrotizing pancreatitis). The therapeutic intervention plan should be crafted to reverse signs and symptoms, restore normocalcemia, and correct or manage the underlying cause of hypercalcemia.
General Approach to Treatment
Chronic hypercalcemia is usually caused by an underlying medical condition or prescribed pharmacotherapies that can be resolved by successful treatment of the condition or withdrawal of the offending agent. Acute hypercalcemic episodes induced by malignancies may be mitigated by chemotherapy and/or radiation treatment. Effective surgical or drug treatment of primary hyperparathyroidism should reduce serum calcium concentrations as well as reduce the development of long-term complications such as vascular complications, chronic kidney disease (CKD), and kidney stones.
Nonpharmacologic Therapy
Hypercalcemic crisis and acute symptomatic severe hypercalcemia should be considered medical emergencies and treated immediately (Fig. 35-2).
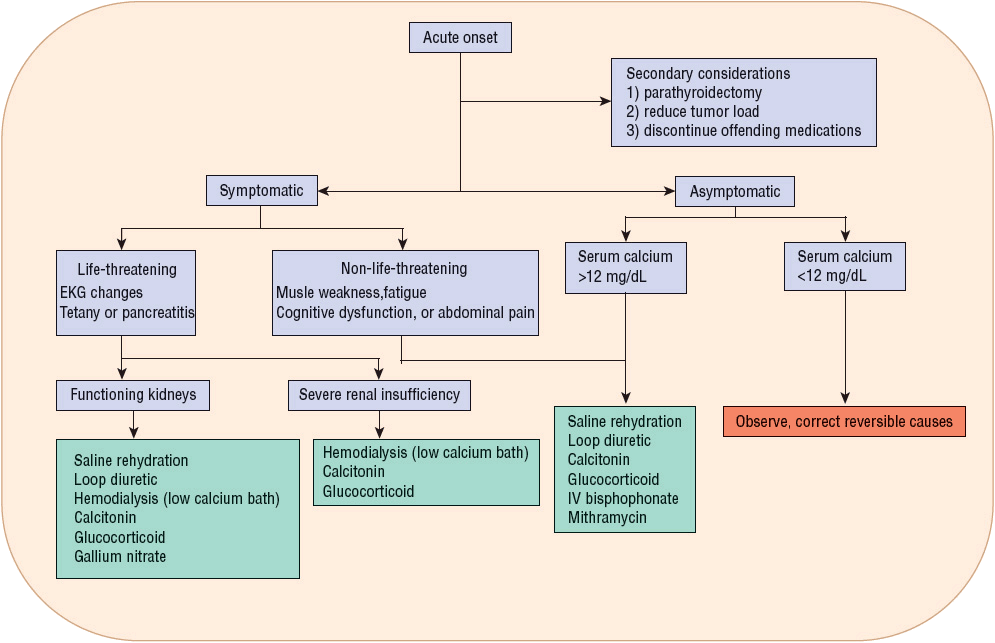
FIGURE 35-2 Pharmacotherapeutic options for the acutely hypercalcemic patient. Serum calcium of 12 mg/dL is equivalent to 3 mmol/L.
These patients may require immediate-acting interventions to promptly reduce the serum calcium concentration if they are experiencing ECG changes, neurologic manifestations, or pancreatitis. Pharmacologic therapy consisting of volume expansion and enhancement of urinary calcium excretion with loop diuretics is usually the initial management strategy. Hemodialysis against a zero- or low-calcium dialysate solution should be considered for patients with severely impaired renal function (CKD stage 4 or 5) who cannot tolerate large fluid loads and in whom diuretics have limited efficacy.22
Effective treatment of moderate to severe hypercalcemia in the absence of life-threatening symptoms begins with attention to the underlying disorder and correction of associated fluid and electrolyte abnormalities. Patients with primary hyperparathyroidism may require surgery, particularly if they have systemic manifestations.
Clinical Controversy…
Patients with malignancy often require surgical or chemotherapeutic reduction of tumor load to control the exogenous supply of cytokines and hormones (e.g., PTHrP) that cause hypercalcemia. In contrast, patients with drug-induced hypercalcemia generally respond to discontinuation of the offending agent.23
Pharmacologic Therapy
![]() For those patients with normal to moderately impaired renal function (CKD stages 3 and 4), the cornerstone of initial treatment of severe hypercalcemia or hypercalcemic crisis is volume expansion with normal saline to increase natriuresis and ultimately urinary calcium excretion (see Table 35-2). Patients with symptomatic hypercalcemia are often extracellular volume depleted secondary to vomiting and polyuria; thus rehydration with saline-containing fluids is necessary to interrupt the stimulus for sodium and calcium reabsorption in the renal tubule.24 Rehydration can be accomplished by the infusion of normal saline at rates of 200 to 300 mL/h, until the patient is fluid resuscitated and serum calcium approaches the upper limit of the normal range. The precise rate depends on concomitant conditions (primarily cardiovascular and renal) and magnitude of hypercalcemia. The saline infusion rate can be decreased to a rate that approximates the patient’s intake of oral or IV fluids. (see Chap. 34 for a thorough discussion of how to calculate water deficit.) Adequacy of hydration is assessed by measuring fluid intake and output or by central venous pressure monitoring.9 Loop diuretics such as furosemide (40 to 80 mg IV every 1 to 4 hours) or ethacrynic acid (for patients with sulfa allergies) can also be instituted to increase urinary calcium excretion and to minimize the development of volume overload from the administration of saline9 (Fig. 35-2 and Table 35-2). Loop diuretics such as furosemide block calcium (and sodium) reabsorption in the thick ascending limb of the loop of Henle and augment the calciuric effect of saline alone. The importance of rehydration prior to loop diuretic use is critical because if dehydration persists or becomes worse, the serum calcium can actually increase because of enhanced proximal tubule calcium reabsorption.2 Potassium chloride, 10 to 20 mEq/L (10 to 20 mmol/L), should be added to the saline solution after rehydration is accomplished to maintain normokalemia in the presence of diuretic therapy. Serum magnesium levels should also be monitored, and magnesium replacement instituted if magnesium levels fall below 1.8 mg/dL (0.74 mmol/L) (Table 35-3). Rehydration with saline and administration of furosemide can result in a decrease of 2 to 3 mg/dL (0.50 to 0.75 mmol/L) in total serum calcium within 24 to 48 hours.9
For those patients with normal to moderately impaired renal function (CKD stages 3 and 4), the cornerstone of initial treatment of severe hypercalcemia or hypercalcemic crisis is volume expansion with normal saline to increase natriuresis and ultimately urinary calcium excretion (see Table 35-2). Patients with symptomatic hypercalcemia are often extracellular volume depleted secondary to vomiting and polyuria; thus rehydration with saline-containing fluids is necessary to interrupt the stimulus for sodium and calcium reabsorption in the renal tubule.24 Rehydration can be accomplished by the infusion of normal saline at rates of 200 to 300 mL/h, until the patient is fluid resuscitated and serum calcium approaches the upper limit of the normal range. The precise rate depends on concomitant conditions (primarily cardiovascular and renal) and magnitude of hypercalcemia. The saline infusion rate can be decreased to a rate that approximates the patient’s intake of oral or IV fluids. (see Chap. 34 for a thorough discussion of how to calculate water deficit.) Adequacy of hydration is assessed by measuring fluid intake and output or by central venous pressure monitoring.9 Loop diuretics such as furosemide (40 to 80 mg IV every 1 to 4 hours) or ethacrynic acid (for patients with sulfa allergies) can also be instituted to increase urinary calcium excretion and to minimize the development of volume overload from the administration of saline9 (Fig. 35-2 and Table 35-2). Loop diuretics such as furosemide block calcium (and sodium) reabsorption in the thick ascending limb of the loop of Henle and augment the calciuric effect of saline alone. The importance of rehydration prior to loop diuretic use is critical because if dehydration persists or becomes worse, the serum calcium can actually increase because of enhanced proximal tubule calcium reabsorption.2 Potassium chloride, 10 to 20 mEq/L (10 to 20 mmol/L), should be added to the saline solution after rehydration is accomplished to maintain normokalemia in the presence of diuretic therapy. Serum magnesium levels should also be monitored, and magnesium replacement instituted if magnesium levels fall below 1.8 mg/dL (0.74 mmol/L) (Table 35-3). Rehydration with saline and administration of furosemide can result in a decrease of 2 to 3 mg/dL (0.50 to 0.75 mmol/L) in total serum calcium within 24 to 48 hours.9
TABLE 35-2 Drug Dosing Table for Hypercalcemia
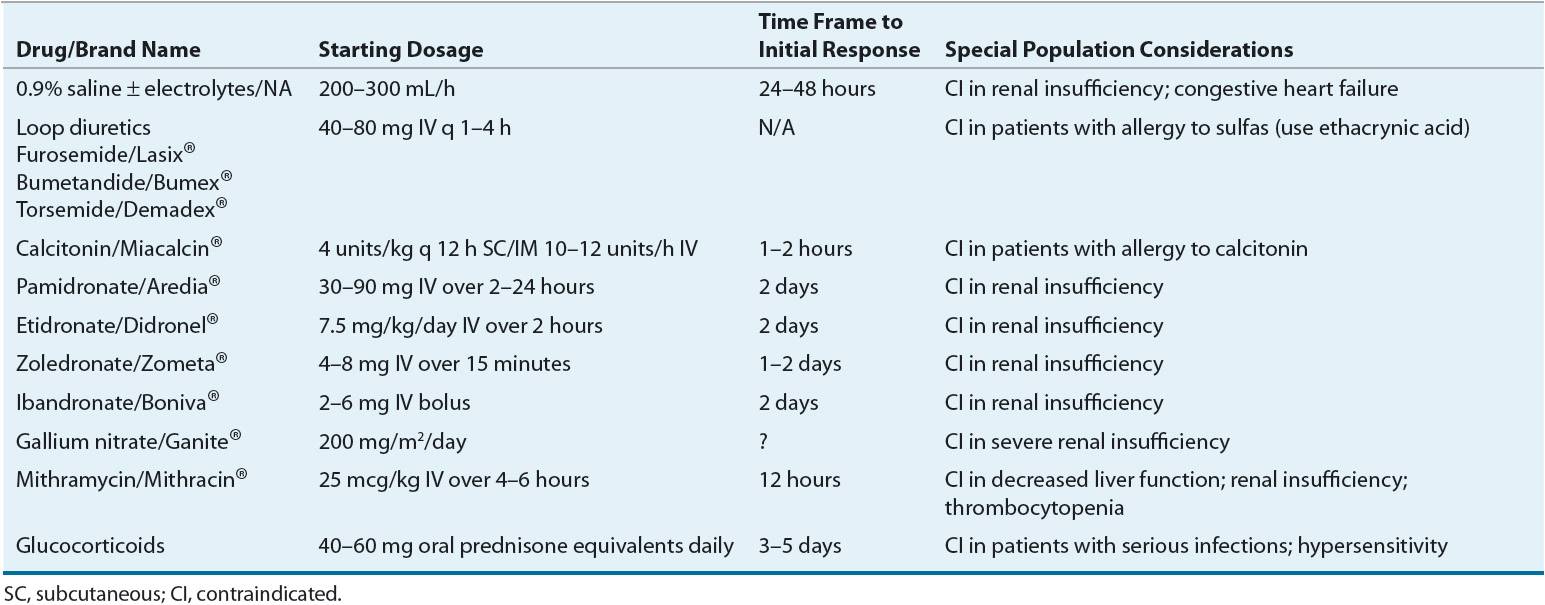
TABLE 35-3 Hypercalcemia Drug Monitoring Table
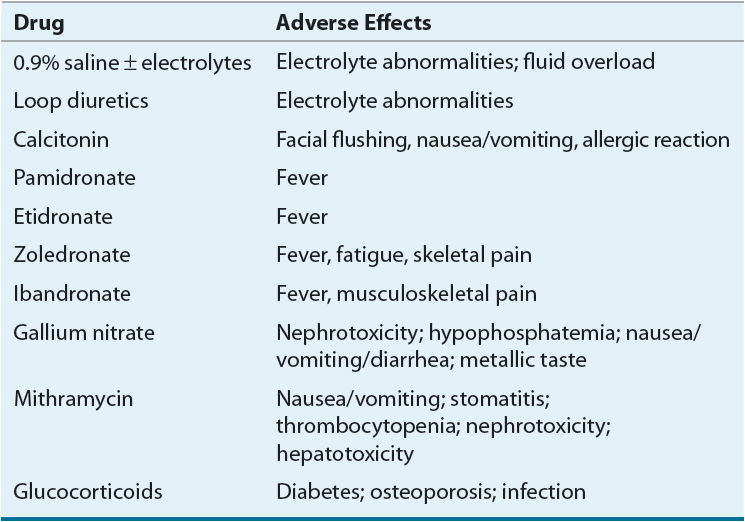
Alternative Drug Treatments
Calcitonin
In those patients in whom saline hydration therapy is contraindicated (e.g., those with severe chronic heart failure [CHF] or moderate-to-severe renal dysfunction), short-term therapy with calcitonin is a viable alternative agent to initiate reduction of serum calcium levels within 24 to 48 hours. Calcitonin has a rapid onset of action (within 1 to 2 hours); however, the degree and extent of serum calcium level reduction are often unpredictable.2
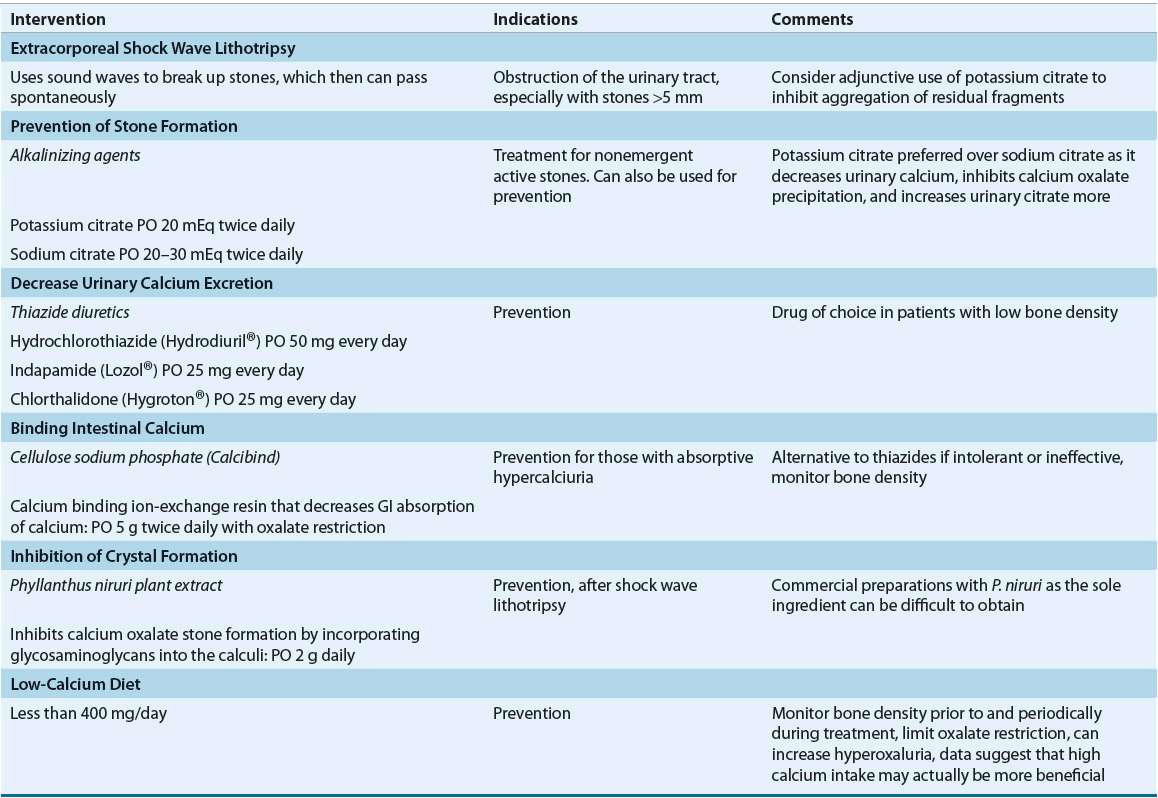
Subcutaneous administration of salmon calcitonin, 50 to 100 international units daily or three times weekly, has been used to manage mild hypercalcemia in patients with Paget’s disease.25 The intranasal formulation of calcitonin has been used in doses of 200 to 400 international units daily; unfortunately, this has resulted in only mild decreases in serum calcium. The lack of significant efficacy of the synthetic intranasal formulation is the result of the lower potency and shorter duration of action as compared to salmon calcitonin. Patients who develop nephrolithiasis from hypercalciuria are most often treated with sodium citrate to prevent stone formation, thiazide diuretics to decrease urinary calcium excretion, or shock wave lithotripsy (Table 35-4).
TABLE 35-4 Treatment of Nephrolithiasis Associated with Chronic Hypercalcemia and Hypercalciuria
Pharmacology Calcitonin decreases serum calcium concentrations, primarily by inhibiting bone resorption. It can also reduce renal tubular reabsorption of calcium, thus promoting calciuresis.26 Recently, the calcitonin receptor has been shown to play an important role in calcium homeostasis, particularly in states of calcium stress (e.g., vitamin D toxicity).26 Calcitonin from salmon sources is most commonly administered subcutaneously or intramuscularly (for larger volumes) in a starting dose of 4 units/kg every 12 hours.
Adverse Effects The side effects from IV administered calcitonin (facial flushing, nausea, and vomiting) limit patient acceptability. Allergic reactions, although rare, do occur; therefore, a test dose (intradermal injection of 0.1 mL of a 10 units/mL solution) is recommended prior to starting therapy. If marked erythema and/or wheal formation does not occur within 15 minutes after administration, therapy can begin. Salmon calcitonin therapy is associated with tachyphylaxis caused by antibody formation to foreign proteins or molecules resembling the calcitonin polypeptide.26 Tachyphylaxis has been primarily documented in patients receiving therapy for more than 4 months and thus might not be clinically significant in the acute care setting. The addition of corticosteroid therapy or conversion to human calcitonin increases effectiveness.2
Bisphosphonates
Bisphosphonates block bone resorption very efficiently, render the hydroxyapatite crystal of bone mineral resistant to hydrolysis by phosphatases, and also inhibit osteoclast precursors from attaching to the mineralized matrix, thus blocking their transformation into mature functioning osteoclasts.15,27 The antiresorptive properties of this class of agents can provide long-term control of serum calcium and are the first-line therapy for cancer-associated hypercalcemia.
Pharmacology and Dosing Pamidronate is very effective in controlling hypercalcemia associated with malignancy and slightly more effective than etidronate.7 The usual dose of pamidronate is 30 to 90 mg as an IV infusion given over 2 to 24 hours. Pamidronate also has the advantage of single-day therapy.9 Etidronate, when administered in doses of 7.5 mg/kg per day by slow IV infusion over at least 2 hours for 3 days, is effective in the therapy of hypercalcemia of malignancy.9 Zoledronate and ibandronate are newer, high-potency bisphosphonates with demonstrated effectiveness in the treatment of hypercalcemia of malignancy. Complete response has been reported in 88.4% to 86.7% of zoledronate- versus 69.7% of pamidronate-treated patients.28,29 Zoledronate IV doses of 4 to 8 mg given over 5 minutes have resulted in normalization of serum calcium concentrations.29 IV infusions of 0.02 or 0.04 mg/kg diluted in 5% dextrose (given over 20 to 50 minutes) have also been effective.30 A similar hypocalcemic response has been noted with ibandronate in comparison with pamidronate (76.5% vs. 75.8%); however, the time period to a relapse of hypercalcemia was longer with ibandronate (14 vs. 4 days), suggesting a therapeutic advantage for ibandronate.31 In contrast to other bisphosphonates, ibandronate can be administered by bolus injection. Single doses of 4 to 6 mg when administered every 3 to 4 weeks have been effective in managing hypercalcemia of malignancy.32 The onset of serum calcium concentration decline is slower with bisphosphonate therapy (concentrations begin to decline in 2 days and reach a nadir in 7 days); thus calcitonin therapy can be necessary if rapid serum level reduction is required.9,32 Duration of normocalcemia varies, but usually does not exceed 2 to 3 weeks. It appears to be dependent on the severity and treatment response of the underlying malignancy.2 The duration of response has been suggested to be longer with zoledronate (4 to 5 weeks), although the data are sparse.30
Adverse Effects Fever is a common side effect of IV bisphosphonate therapy. Although oral bisphosphonates are useful for the treatment of bone turnover in Paget’s disease, there are insufficient data to suggest their use for the initial treatment of hypercalcemia. The use of oral bisphosphonates for maintenance therapy in patients predisposed to hypercalcemia (malignancy) has been successful in some cases.33 The safety of continuous bisphosphonate therapy in patients with moderate-to-severe renal insufficiency is currently unknown. Renal function monitoring (serum creatinine) is advised with the use of bisphosphonates, as cases of acute tubular necrosis have been reported.34,35 Although there are no published guidelines for frequency of serum creatinine monitoring, it is advisable to evaluate serum creatinine within a week after the infusion and just prior to the next scheduled dose.
Denosumab
Denosumab is a monoclonal antibody that inhibits the receptor activator of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) ligand (RANKL), a principal mediator of osteoclast survival. Denosumab is FDA approved for the treatment of osteoporosis.
Denosumab has been investigated in patients with malignancies and bone metastases (without hypocalcemia) who were either bisphosphonate naïve or who had previous exposure to bisphosphonates. Patients were randomized to receive an IV bisphosphonate every 4 weeks (91% received zoledronic acid) or a fixed dose of denosumab every 4 weeks (30, 120, and 180 mg) or every 12 weeks (60 and 180 mg) to investigate the effect on the primary outcome measure of urinary-N-telopeptide, a marker for bone turnover. Patients who were bisphosphonate naïve had similar decline in markers of bone turnover. In patients with previous exposure to bisphosphonates, treatment with denosumab produced significantly greater reductions in bone turnover compared with an IV bisphosphonate.36 A recent clinical trial in breast cancer patients that were randomized to subcutaneous denosumab 120 mg or IV zolendronic acid 4 mg and placebo every 4 weeks showed that denosumab therapy prolonged time to first skeletal-related event or hypercalcemia by 18%.37 These data indicate utility of denosumab in hypercalcemia of malignancy, particularly in patients who do not have an optimal response to bisphosphonates.38 Denosumab has also been reported to successfully treat hypercalcemia after successful stem cell transplantation and restitution of osteoclast function in patients with osteopetrosis, a heritable disorder associated with defective osteoclast function.39
Gallium Nitrate
Gallium nitrate is indicated for the treatment of symptomatic hypercalcemia of malignancy not responsive to hydration therapy.40 However, because of its adverse side-effect profile, it is generally reserved for those who fail to respond to less toxic agents. Gallium nitrate inhibits bone resorption, and may be superior to calcitonin in inducing normocalcemia. The initial dose is usually a continuous IV infusion of 200 mg/m2 per day for five consecutive days. Gallium nitrate can be more effective in achieving normocalcemia in patients with epidermoid (squamous) cancers.41 Because gallium nitrate is nephrotoxic, the initial dose should be conservative and the patient’s renal function should be closely monitored.
Mithramycin
Mithramycin (plicamycin) is a potent cytotoxic antibiotic that inhibits osteoclast-mediated bone resorption and thereby reduces hypercalcemia. Mithramycin can be administered via IV infusion (25 mcg/kg) over 4 to 6 hours in saline or 5% dextrose solutions. This therapy can be repeated daily for 3 to 4 days or on alternating days for three to eight doses.9,42 Serum calcium levels begin to fall within 12 hours of a mithramycin dose, with the peak effect generally occurring over 48 to 96 hours.2,9 Common dose-related adverse effects of mithramycin include nausea, vomiting, stomatitis, thrombocytopenia, inhibition of platelet function, and renal and hepatotoxicity.
Corticosteroids
Prednisone or an equivalent agent is usually effective in the treatment of hypercalcemia resulting from multiple myeloma, leukemia, lymphoma, sarcoidosis, and hypervitaminoses A and D.2,27,42 These agents are effective because they reduce GI calcium absorption.42 Corticosteroids may also prevent tachyphylaxis to salmon calcitonin.26 Daily doses of 40 to 60 mg of prednisone or the equivalent are effective at reducing serum calcium within 3 to 5 days followed by a reduction in urinary calcium excretion within 7 to 10 days. The disadvantages of corticosteroid therapy are its relatively slow onset of action and the potential for diabetes mellitus, osteoporosis, and increased susceptibility to infection.43
Cinacalcet
The calcimimetic agent cinacalcet HCl is approved for management of parathyroid carcinoma.44,45 It binds to the CaSr, and increases the sensitivity for receptor activation by extracellular calcium. This results in reduced PTH and serum calcium concentrations.44,45 Cinacalcet HCl administered at a starting dose of 30 mg orally twice daily has been used for the treatment of hypercalcemia secondary to parathyroid carcinoma. The dosage is titrated every 2 to 4 weeks in 30-mg increments until the desired serum calcium level is achieved. The maximum approved dosage is 90 mg three to four times daily. Patients should have serum calcium measured within 1 week after starting or increasing the dose of this agent.46
HYPOCALCEMIA
![]() Hypocalcemia occurs infrequently in the outpatient setting and is most common in elderly, malnourished patients and those who have received sodium phosphate as a bowel preparation agent.
Hypocalcemia occurs infrequently in the outpatient setting and is most common in elderly, malnourished patients and those who have received sodium phosphate as a bowel preparation agent.
Incidence
The incidence of hypocalcemia in intensive care unit patients ranges from 70% to 90% based on total serum calcium values less than 8.5 mg/dL (<2.13 mmol/L) to 15% to 50% based on the observation of ionized calcium concentrations less than 4.4 mg/dL (<1.10 mmol/L).3 Emergent treatment of hypocalcemia is rarely warranted unless life-threatening symptoms are present (e.g., frank tetany or seizures).
Pathophysiology
Hypocalcemia is the result of alterations in the effect of PTH and vitamin D on the bone, gut, and kidney (see Fig. 35-1). The primary causes of hypocalcemia are postoperative hypoparathyroidism and vitamin D deficiency. Other causes include magnesium deficiency, thyroid surgery, medications, hypoalbuminemia, blood transfusions, peripheral blood progenitor cell harvesting, tumor lysis syndrome, and mutations in the CaSr.47–52 PTH concentrations are elevated in conditions of hypocalcemia, with the exception of hypoparathyroidism and hypomagnesemia.53
Vitamin D Deficiency
Vitamin D and its metabolites play an important role in the maintenance of extracellular calcium concentrations and in normal skeletal structure and mineralization. Vitamin D is necessary for the optimal absorption of calcium and phosphorus. On a worldwide basis, the most common cause of chronic hypocalcemia is nutritional vitamin D deficiency. In malnourished populations, manifestations include rickets and osteomalacia. Nutritional vitamin D deficiency is uncommon in Western societies because of the fortification of milk with ergocalciferol. The most common cause of vitamin D deficiency in Western societies is GI disease.15 Gastric surgery, chronic pancreatitis, small-bowel disease, intestinal resection, and bypass surgery are associated with decreased concentrations of vitamin D and its metabolites.15 Vitamin D replacement therapy might need to be administered by the IV route if poor oral bioavailability is noted. Decreased production of 1,25-dihydroxyvitamin D3 can occur as a result of a hereditary defect resulting in vitamin D-dependent rickets.53 Recently, polymorphisms of the vitamin D receptor have been identified, and these genetic variations can contribute to increased risk of rickets associated with vitamin D and calcium deficient diets, especially in certain African and East Asian populations.54 It also can occur secondary to CKD if there is insufficient production of the 1-α-hydroxylase enzyme for the production of the most active metabolite, 1,25-dihydroxy vitamin D3. Treatment of hypocalcemia associated with CKD is reviewed in Chapter 29.
Hypomagnesemia
Hypomagnesemia of any cause can be associated with severe symptomatic hypocalcemia that is unresponsive to calcium replacement therapy (see Chap. 36). Reduced serum magnesium concentrations can impair PTH secretion and induce resistance of target organs to the actions of PTH.15 Normalization of serum calcium concentrations in these patients is thus dependent on appropriate replacement of magnesium.
Hungry Bone Syndrome
An acute, symptomatic rapid fall in total serum calcium concentration (to values <7 mg/dL [<1.75 mmol/L]) is common in patients who have recently had a parathyroidectomy or thyroidectomy. Hypocalcemia in these postsurgical patients is generally transient in nature.53 The “hungry bone syndrome” is a condition of profound hypocalcemia whereby the bone avidly incorporates calcium and phosphorus from the blood in an attempt to recalcify bone.55 Serum calcium concentrations should be monitored every 6 hours during the 24 to 48 hours following such surgeries, and pharmacologic doses of calcium can be necessary to prevent or minimize the drop in serum calcium. Additionally, mild-to-moderate hypocalcemia can be a long-term consequence of parathyroidectomy in hemodialysis patients.53
Drug-Induced Hypocalcemia
Drug-induced hypocalcemia has been reported in patients receiving furosemide, calcitonin, bisphosphonates, gallium nitrate, mithramycin, cinacalcet, fluoride, ketoconazole, and pentamidine.46,56



