The identification of the four genes associated with AD has provided great insight not only into the pathogenesis of monogenic AD but also, as is commonly the case in medical genetics, into the mechanisms that underlie the more common form, nonfamilial or sporadic AD. Indeed, overproduction of one proteolytic product of βAPP, called the Aβ peptide, appears to be at the center of AD pathogenesis, and the currently available experimental evidence suggests that the βAPP, presenilin 1, and presenilin 2 proteins all play a direct role in the pathogenesis of AD.
The Pathogenesis of Alzheimer Disease: β-Amyloid Peptide and Tau Protein Deposits
The most important pathological abnormalities of AD are the deposition in the brain of two fibrillary proteins, β-amyloid peptide (Aβ) and tau protein. The Aβ peptide is generated from the larger βAPP protein (see Table 12-5), as discussed in the next section, and is found in extracellular amyloid or senile plaques in the extracellular space of AD brains. Amyloid plaques contain other proteins besides the Aβ peptide, notably apoE (see Table 12-5). Tau is a microtubule-associated protein expressed abundantly in neurons of the brain. Hyperphosphorylated forms of tau compose the neurofibrillary tangles that, in contrast to the extracellular amyloid plaques, are found within AD neurons. The tau protein normally promotes the assembly and stability of microtubules, functions that are diminished by phosphorylation. Although the formation of tau neurofibrillary tangles appears to be one of the causes of the neuronal degeneration in AD, mutations in the tau gene are associated not with AD but with another autosomal dominant dementia, frontotemporal dementia.
The Amyloid Precursor Protein Gives Rise to the β-Amyloid Peptide
The major features of the βAPP and its corresponding gene are summarized in Table 12-5. βAPP is a single-pass intracellular transmembrane protein found in endosomes, lysosomes, the ER and the Golgi apparatus. It is subject to three distinct proteolytic fates, depending on the relative activity of three different proteases: α-secretase and β-secretase, which are cell surface proteases; and γ-secretase, which is an atypical protease that cleaves membrane proteins within their transmembrane domains. The predominant fate of approximately 90% of βAPP is cleavage by the α-secretase (Fig. 12-24), an event that precludes the formation of the Aβ peptide, because α-secretase cleaves within the Aβ peptide domain. The other approximately 10% of βAPP is cleaved by the β- and γ-secretases to form either the nontoxic Aβ40 peptide or the Aβ42 peptide. The Aβ42 peptide is thought to be neurotoxic because it is more prone to aggregation than its Aβ40 counterpart, a feature that makes AD a conformational disease like α1AT deficiency (described previously in this chapter). Normally, little Aβ42 peptide is produced, and the factors that determine whether γ-secretase cleavage will produce the Aβ40 or Aβ42 peptide are not well defined.
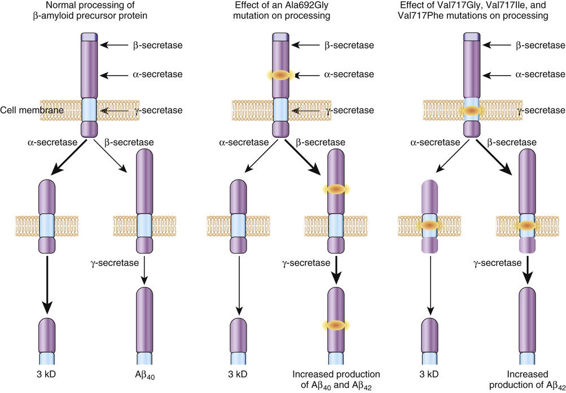
In monogenic AD due to missense substitutions in the gene encoding βAPP (APP), however, several mutations lead to the relative overproduction of the Aβ42 peptide. This increase leads to accumulation of the neurotoxic Aβ42, an occurrence that appears to be the central pathogenic event of all forms of AD, monogenic or sporadic. Consistent with this model is the fact that patients with Down syndrome, who possess three copies of the APP gene (which is on chromosome 21), typically develop the neuropathological changes of AD by 40 years of age. Moreover, mutations in the AD genes presenilin 1 and presenilin 2 (see Table 12-5) also lead to increased production of Aβ42. Notably, the amount of the neurotoxic Aβ42 peptide is increased in the serum of individuals with mutations in the βAPP, presenilin 1, and presenilin 2 genes; furthermore, in cultured cell systems, the expression of mutant βAPP, presenilin 1, and presenilin 2 increases the relative production of Aβ42 peptide by twofold to tenfold.
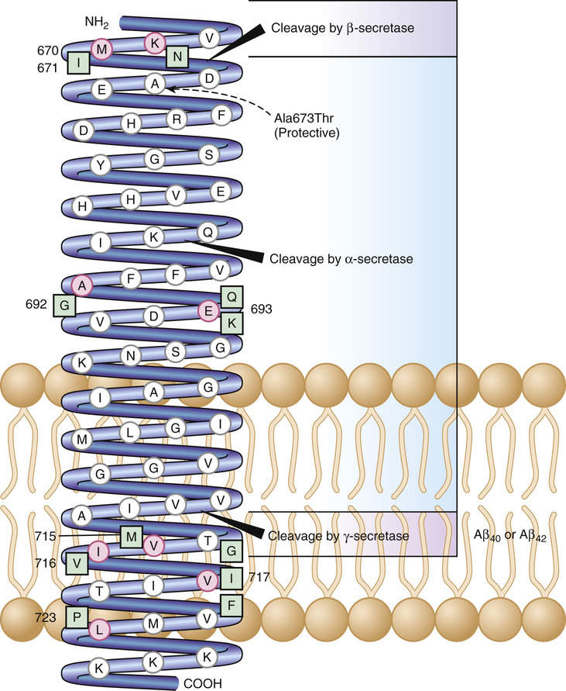
The central role of the Aβ42 peptide in AD is highlighted by the discovery of a coding mutation (Ala673Thr) in the APP gene (Fig. 12-25) that protects against both AD and cognitive decline in older adults. The protective effect is likely due to reduced formation of the Aβ42 peptide, reflecting the proximity of Thr673 to the β-secretase cleavage site (see Fig. 12-25).
The Presenilin 1 and 2 Genes
The genes encoding presenilin 1 and presenilin 2 (see Table 12-5) were identified in families with autosomal dominant AD. Presenilin 1 is required for γ-secretase cleavage of βAPP derivatives. Indeed, some evidence suggests that presenilin 1 is a critical cofactor protein of γ-secretase. The mutations in presenilin 1 associated with AD, through an unclear mechanism, increase production of the Aβ42 peptide. A major difference between presenilin 1 and presenilin 2 mutations is that the age at onset with the latter is much more variable (presenilin 1, 35 to 60 years; presenilin 2, 40 to 85 years); indeed, in one family, an asymptomatic octogenarian carrying a presenilin 2 mutation transmitted the disease to his offspring. The basis of this variation is partly dependent on the number of APOE ε4 alleles (see Table 12-5 and later discussion) carried by individuals with a presenilin 2 mutation; two ε4 alleles are associated with an earlier age at onset than one allele, and one confers an earlier onset than other APOE alleles.
The APOE Gene is an Alzheimer Disease Susceptibility Locus
As presented in Chapter 8, the ε4 allele of the APOE gene is a major risk factor for the development of AD. The role for APOE as a major AD susceptibility locus was suggested by multiple lines of evidence, including linkage to AD in late-onset families, increased association of the ε4 allele with AD patients compared with controls, and the finding that apoE binds to the Aβ peptide. The APOE protein has three common forms encoded by corresponding APOE alleles (Table 12-6). The ε4 allele is significantly overrepresented in patients with AD (≈40% vs. ≈15% in the general population) and is associated with an early onset of AD (for ε4/ε4 homozygotes, the age at onset of AD is approximately 10 to 15 years earlier than in the general population; see Chapter 8). Moreover, the relationship between the ε4 allele and the disease is dose-dependent; two copies of ε4 are associated with an earlier age at onset (mean onset before 70 years) than with one copy (mean onset after 70 years) (see Fig. 8-11 and Table 8-14). In contrast, the ε2 allele has a protective effect and correspondingly is more common in elderly subjects who are unaffected by AD (see Table 12-6).

The mechanisms underlying these effects are not known, but apoE polymorphisms may influence the processing of βAPP and the density of amyloid plaques in AD brains. It is also important to note that the APOE ε4 allele is not only associated with an increased risk for AD; carriers of ε4 alleles can also have poorer neurological outcomes after head injury, stroke, and other neuronal insults. Although carriers of the APOE ε4 allele have a clearly increased risk for development of AD, there is currently no role for screening for the presence of this allele in healthy individuals; such testing has poor positive and negative predictive values and would therefore generate highly uncertain estimates of future risk for AD (see Chapter 18).
Other Genes Associated with AD
One significant modifier of AD risk, the TREM2 gene (which encodes the so-called triggering receptor expressed on myeloid cells 2), was identified by whole-exome and whole-genome sequencing in families with multiple individuals affected with AD. Several moderately rare missense coding variants in this gene are associated with a fivefold increase in risk for late-onset AD, making TREM2 mutations the second most common contributor to classic late-onset AD after APOE ε4. Statistical analyses suggest that an additional four to eight genes may significantly modify the risk for AD, but their identity remains obscure.
Although case-control association studies (see Chapter 10) of candidate genes with hypothetical functional links to the known biology of AD have suggested more than 100 genes in AD, only one such candidate gene, SORL1 (sortilin-related receptor 1), has been robustly implicated. Single nucleotide polymorphisms (SNPs) in the SORL1 gene confer a moderately increased relative risk for AD of less than 1.5. The SORL1-encoded protein affects the processing of APP and favors the production of the neurotoxic Aβ42 peptide from βAPP.
Genome-wide association studies analyses (see Chapter 10), on the other hand, have greatly expanded the number of genes believed to be associated with AD, identifying at least nine novel SNPs associated with a predisposition to nonfamilial late-onset forms of AD. The genes implicated by these SNPs and their causal role(s) in AD are presently uncertain.
Overall, it is becoming clear that genetic variants alter the risk for AD in at least two general ways: first, by modulating the production of Aβ, and second, through their impact on other processes, including the regulation of innate immunity, inflammation, and the resecretion of protein aggregates. These latter variants likely modulate AD risk by altering the flux through downstream pathways in response to a given load of Aβ.
Diseases of Mitochondrial DNA (mtDNA)
The mtDNA Genome and the Genetics of mtDNA Diseases
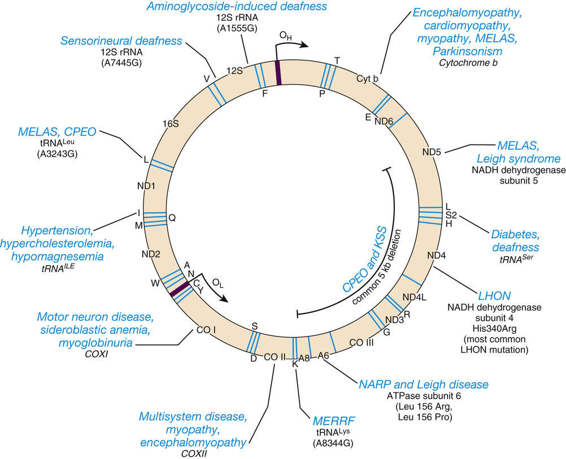
The general characteristics of the mtDNA genome and the features of the inheritance of disorders caused by mutations in this genome were first described in Chapters 2 and 7 but are reviewed briefly here. The small circular mtDNA chromosome is located inside mitochondria and contains only 37 genes (Fig. 12-26). Most cells have at least 1000 mtDNA molecules, distributed among hundreds of individual mitochondria, with multiple copies of mtDNA per mitochondrion. In addition to encoding two types of ribosomal RNA (rRNA) and 22 transfer RNAs (tRNAs), mtDNA encodes 13 proteins that are subunits of oxidative phosphorylation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


