Learning Objectives
Learn the differential diagnosis of leukopenia.
Distinguish between neoplastic and nonneoplastic proliferations of white blood cells.
Learn the diagnostic criteria for the different types of lymphomas, leukemias, myelodysplastic syndromes, myeloproliferative disorders, and plasma cell dyscrasias.
Understand the genetic, biochemical, and/or cellular defects associated with the more commonly encountered disorders of WBC function.
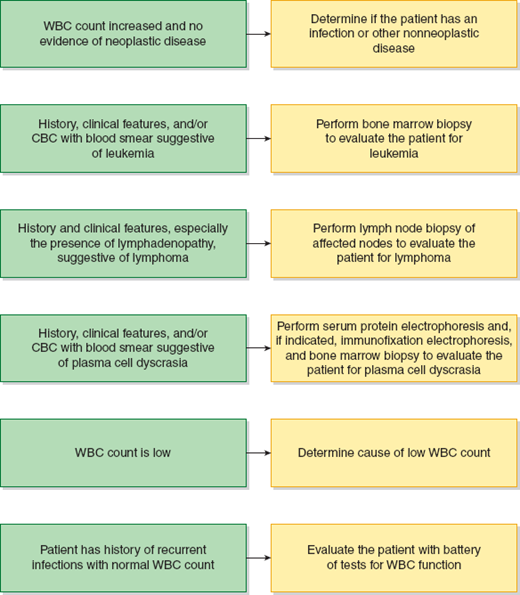
A bnormalities in white blood cells (WBCs) are nearly always quantitative (eg, too many or too few WBCs). These disorders may be neoplastic, as found in leukemia, or nonneoplastic. A qualitative or functional disorder of WBCs may accompany the quantitative disorder. Qualitative defects in WBC function with a normal WBC count occur, but they are uncommon. The approach to diagnosis of WBC disorders is shown in Figure 13–1.
Leukopenia
A low WBC count can occur because of a decreased number of lymphocytes, granulocytes, or both. A number of the immunodeficiency diseases are associated with a lymphocytopenia (see Chapter 3). Granulocytopenia primarily reflects a reduction in the number of neutrophils (neutropenia) in the peripheral blood. When the number of neutrophils decreases below about 1000 neutrophils/μL, the neutropenic patient becomes susceptible to infections. These illnesses range from mild to severe, depending on the type of organism and the effectiveness of the antibiotics used to treat it. A classification of granulocytopenic disorders follows.
A low WBC count can occur because of a decreased number of lymphocytes, granulocytes, or both.
Defects in the production of granulocytes may be caused by:
Diseases associated with marrow failure, such as aplastic anemia.
Diseases in which the marrow is infiltrated by leukemic cells or by metastatic cancer cells originating from another site; the decreased neutrophil production in this setting is typically associated with defects in the production of other blood cells as well.
Suppression of granulocyte production by exposure to certain drugs; the list of drugs that can produce neutropenia is extensive; noteworthy examples are chemotherapeutic agents used in cancer treatment and certain nonsteroidal anti-inflammatory drugs (NSAIDs).
Vitamin B12 or folate deficiency; these disorders produce a megaloblastic anemia and defective DNA synthesis in granulocyte precursors.
Suppression of granulocyte production by neoplastic cells, for example, large granular lymphocytic leukemia.
Accelerated removal of granulocytes may be caused by:
Immunologically mediated injury to neutrophils following exposure to drugs, with the injury occurring from an immune response on the neutrophil surface.
Immunologically mediated injury to neutrophils as part of an autoimmune disorder; for example, Felty syndrome is a variant of rheumatoid arthritis with neutropenia, splenomegaly, leg ulcers, and the joint lesions found in rheumatoid arthritis; the neutropenia can dominate the clinical course in patients with Felty syndrome.
Immunologically mediated injury to neutrophils that is idiopathic and not associated with any identifiable abnormality.
Excessive destruction of granulocytes from splenic sequestration of the neutrophils in an enlarged spleen or from overwhelming infection.
Nonneoplastic Proliferation of WBCs
An elevated peripheral WBC count is commonly found in patients with infections and other inflammatory states, such as those associated with autoimmune disorders.
An elevated peripheral WBC count is commonly found in patients with infections and other inflammatory states, such as those associated with autoimmune disorders.
Patients can develop a lymphocytosis in a variety of different conditions such as tuberculosis, acute bowel infections, and infectious mononucleosis and other viral infections.
An increase in circulating eosinophils is most commonly found in patients with allergic disorders and those with asthma. An increase in circulating eosinophils is also found in patients with certain parasitic infections and in patients with dermatologic disorders such as eczema. Increases in eosinophils can also be caused by some drugs and some autoimmune disorders. Finally, increases in eosinophils can be seen in certain neoplastic conditions such as Hodgkin lymphoma and T-cell lymphomas.
The peripheral monocyte count is increased in a number of situations where the lymphocyte count is also increased, such as tuberculosis. Rheumatoid arthritis, systemic lupus erythematosus, and other connective tissue diseases also may be associated with a monocytosis.
A mild increase in circulating neutrophils can occur without disease after strenuous exercise, during menstruation, and in the course of pregnancy. An increased neutrophil count is clinically significant when it is indicative of a bacterial infection, a neoplastic disorder, ischemia, an autoimmune disorder, or an effect of certain drugs, such as corticosteroids or epinephrine. The most frequently identified immature neutrophil in the blood when there is an increased WBC count is the neutrophilic band cell. The percentage of WBCs represented by band cells or more immature neutrophil precursors is a commonly used indicator of infection. However, band counts are poorly reproducible among medical technologists, so the current trend is to not report band counts. Other less mature neutrophil precursors can be seen in infections and other conditions where the bone marrow is attempting to produce granulocytes rapidly.
Neoplastic Proliferation of WBCs
WBC neoplasms frequently involve the peripheral blood, and can result in leukocytosis. White cell neoplasms are broadly divided into 2 large categories, lymphoid (the lymphocyte lineage) and myeloid (the lineage including granulocytes, monocytes, megakaryocytes, and erythroid cells). Lymphoid disorders include acute precursor lymphoblastic leukemias (ALL) and mature B-, T-, and NK-cell neoplasms. Myeloid disorders include acute myeloid leukemias, myeloproliferative neoplasms, and myelodysplastic syndromes.
White cell neoplasms are broadly divided into 2 large categories, lymphoid (the lymphocyte lineage) and myeloid (the lineage including granulocytes, monocytes, megakaryocytes, and erythroid cells).
Lymphoid Malignancies
The lymphoid malignancies are caused by neoplastic transformation of lymphocytes or their precursors. Lymphoid cells can be found in the lymph nodes, blood, bone marrow, spleen, and extranodal sites such as the skin, mucosae, and respiratory and gastrointestinal tracts. Lymphoid neoplasms can occur at any of these sites. Neoplasms that primarily involve the bone marrow and peripheral blood are referred to as leukemias, and those involving tissue sites are called lymphomas. However, many lymphoid malignancies can involve both tissues and the blood/bone marrow, so the leukemia/lymphoma distinction is somewhat arbitrary. The World Health Organization (WHO) classification system for lymphoid malignancies is shown in Table 13–1.
|
|
|
|
|
|
|
|
Lymphoid leukemias can correspond to precursor B or T cells or mature lymphoid cells. Precursor lymphoid malignances are also called lymphoblastic leukemias/lymphomas. Relatively common B- and T-cell malignancies that frequently present in a leukemic phase include chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, hairy cell leukemia, mantle cell lymphoma, Burkitt lymphoma/leukemia, T-cell prolymphocytic leukemia, T-cell large granular lymphocytic leukemia, and Sézary syndrome. Lymphoid leukemias generally present with elevated white counts (specifically lymphocytosis), and depending on the degree of bone marrow involvement, there can be decreased numbers of normal white cells, red cells, and/or platelets. Processes that involve the marrow extensively can result in the presence of myeloid and erythroid precursor cells in the peripheral blood. Leukemias are diagnosed by examination of the peripheral blood smear and bone marrow aspirates and biopsies. Additional studies such as immunophenotyping by flow cytometry, molecular diagnostic techniques, and cytogenetics are frequently used to establish a diagnosis.
When lymphoid malignancies are mostly tissue-based, they are referred to as lymphomas. Lymphomas are monoclonal, neoplastic proliferations of B, T, or NK cells. Most lymphomas are malignancies of mature lymphocytes, but precursor lymphocytic malignances can involve tissues, and thus be classified as lymphomas. Lymphomas are divided into 2 major groups, Hodgkin lymphoma and a much larger variety of lymphomas known generically as non-Hodgkin lymphomas. The patient with lymphoma often presents with an isolated, enlarged superficial lymph node, which may be discovered accidentally on physical exam. Alternatively, the patient may have generalized lymphadenopathy. If the enlarged lymph node develops in a site where it can produce signs and symptoms, it is more likely to be discovered early in the course of disease. An example is the enlargement of lymph nodes in the mediastinum, which can impair blood flow through the large vessels in the chest and produce symptoms on that basis. In some cases, organ involvement may be the first manifestation of a lymphoma. Non-Hodgkin lymphomas, for example, may become symptomatic when there is cellular proliferation in the orbit, the gastrointestinal tract, or the skin. Involvement of the bone marrow and peripheral blood also may be an initial indicator of the presence of a lymphoma.
Lymph node biopsy is the preferred method for diagnosis of lymphoma, since it allows the pathologist to determine the overall tissue architecture and get a large sample of the cells present. Because the lymphoma may not be distributed evenly in all lymph nodes, it may be necessary to biopsy several lymph nodes to establish the diagnosis. In recent years, both fine needle aspiration and biopsy have been used more commonly to make diagnoses of lymphoma. Although fine needle aspiration does not allow optimal evaluation of tissue architecture, diagnostic procedures such as flow cytometry and/or molecular techniques can be used to render a diagnosis on minimal amounts of material.
Principal differentiating factors between Hodgkin and non-Hodgkin lymphomas are:
Hodgkin lymphoma:
(a) Proliferation of cells is typically localized to a single group of nodes such as the cervical or mediastinal nodes.
(b) Proliferating cells spread by contiguity.
(c) Mesenteric lymph nodes are rarely involved.
Non-Hodgkin lymphomas:
(a) Frequent involvement of multiple groups of nodes.
(b) Proliferating cells spread widely and noncontiguously.
(c) Mesenteric lymph nodes are commonly involved.
The Hodgkin and non-Hodgkin lymphomas are classified into clinical stages based on the distribution of the disease. These stages, with increased clinical severity associated with higher stage numbers, are as follows:
Stage I—involvement of 1 group of lymph nodes or 2 contiguous lymph node clusters on the same side of the diaphragm.
Stage II—involvement of 2 or more noncontiguous lymph node groups on the same side of the diaphragm.
Stage III—lymph node involvement above and below the diaphragm; if there is involvement of the spleen, the lymphoma is classified as III(s); and if there is visceral involvement by direct extension, it is known as stage III(e).
Stage IV—widespread disease, often involving the liver, bone marrow, lungs, bones, and skin.
In addition to the above staging scheme, the designation “B” is added for patients who have constitutional symptoms such as fever, night sweats, and weight loss. For example, a patient with involvement of 2 groups of lymph nodes on the same side of the diaphragm with fevers and night sweats would be considered stage IIB. In general, the presence of these “B symptoms” portends a more advanced stage of disease with worse prognosis. In addition, the designation “E” is used to designate lymphomas involving extranodal sites only (eg, the gastrointestinal tract).
Historically the diagnosis of Hodgkin and non-Hodgkin lymphoma was primarily based on the histological appearance of the lymph nodes. For Hodgkin lymphoma, the Rye classification system was used for decades and has now been incorporated with relatively few changes into the current WHO classification system for hematologic malignancies. Classification of non-Hodgkin lymphomas has been more problematic. Non-Hodgkin lymphomas were organized in the Rappaport classification in 1966, the Lukes–Collins classification in 1973-1974, and in 1982 they were reclassified according to the Working Formulation of Clinical Usage by an international panel of experts.
For Hodgkin lymphoma, the Rye classification system was used for decades and has now been incorporated with relatively few changes into the current WHO classification system for hematologic malignancies. Classification of non-Hodgkin lymphomas has been more problematic.
By the early 1990s, significant progress was made in understanding the biology of lymphomas, so newer classification systems were developed based on typing lymphomas with antibodies specific for cytoplasmic and cell surface proteins (immunohistochemistry and flow cytometry) and by detecting specific molecular lesions. In 1994, the REAL classification was introduced by the International Lymphoma Study Group. The goal of the new classification was to integrate morphological, immunologic, and genetic information to better define the disease entities. The REAL classification system was modified somewhat to form the basis for the current (2008) WHO classification system (Table 13–1).
The WHO classification system, like the REAL classification system preceding it, attempts to classify non-Hodgkin lymphomas according to the normal cell equivalent of the neoplastic cells. First, neoplastic cells are classified based on whether they are of B-cell or T-cell/NK-cell origin. Next, the cells are classified by the stage of differentiation to which they correspond. Most B- and T-cell neoplasms correspond to mature B and T cells.
In the end, lymphoma classification is determined by the architectural features observed under the microscope (eg, follicular vs diffuse growth pattern and the microscopic appearance of the malignant cells), the spectrum of proteins expressed on the surfaces and in the cytoplasm of the malignant cells (eg, T- or B-cell markers and proteins not expressed in normal lymphocytes), the presence of clonal rearrangements of the immunoglobulin or T-cell receptor genes, and, in some cases, the presence of specific genetic lesions in the malignant cells. The techniques used for lymphoma diagnosis include light microscopy, immunohistochemistry, flow cytometry, and molecular techniques including cytogenetics, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), and newer techniques such as microarrays.
Since it is beyond the scope of this chapter to discuss all the lymphoid malignancies in detail, the more common disorders have been selected for inclusion.
Neoplasms of immature B and T cells most commonly present as leukemias, with extensive blood and bone marrow involvement, but they can also involve the lymphoid tissues as lymphomas. For example, precursor T-cell leukemia/lymphoma often presents with a mediastinal mass and may not demonstrate blood or bone marrow involvement. ALL accounts for almost one third of all childhood cancers and represents 75% of all pediatric leukemias. The median age at diagnosis is 10 years with a slight male predominance of 1.4:1. Pediatric leukemias are almost always (80%-85%) of a precursor B-cell lineage, with the remainder being T-cell lineage.
ALL accounts for almost one third of all childhood cancers and represents 75% of all pediatric leukemias.
Morphology—Morphologically, the involved tissues show monomorphic collections of medium-sized cells with fine chromatin, high nuclear:cytoplasmic ratios, and inconspicuous nucleoli.
Immunophenotyping—Depending on lineage, the cells will express B- or T-cell surface proteins. Both B- and T-cell precursor cells contain the enzyme terminal deoxynucleotidyl transferase.
Cytogenetics—There are a number of recurrent chromosomal translocations associated with ALL. Hyperdiploidy with 50 or more chromosomes is a favorable prognostic finding. The presence of the Philadelphia chromosome, t(9;22)(q43;q11), is an adverse prognostic finding.
Chronic Lymphocytic Leukemia
CLL is the most common of the non-Hodgkin lymphomas. The median age at diagnosis is 70 years with a slight male predominance of 1.7:1. The neoplastic cells in CLL are mature B cells. CLL is an indolent disease with a highly variable life expectancy. Transformation to aggressive disease occurs in 5% to 10% of cases at any time during the course of the illness, and is usually a terminal event.
CLL is the most common of the non-Hodgkin lymphomas. CLL is an indolent disease with a highly variable life expectancy.
Morphology—The lymphocytes in CLL are usually small and well differentiated. They are sometimes difficult to distinguish from normal lymphocytes, but they can be identified by their somewhat larger size, coarsely clumped chromatin, and tendency to break apart on peripheral blood smears, forming “smudge cells.” CLL can transform into a high-grade B-cell lymphoma known as Richter syndrome in approximately 3% of B-cell CLL cases. Another type of transformation is to the prolymphocytoid form, where patients can have a very high white count of characteristic prolymphocytes with prominent nucleoli.
Immunophenotyping—The CLL tumor cells express low levels of monoclonal surface IgM and IgD in the majority of cases, surface IgM only in approximately 25% of the cases, and surface IgD, other immunoglobulin isotypes, or no surface immunoglobulin in a small percentage of cases. A characteristic finding in CLL is expression of CD5, which is normally a pan-T-cell antigen, but is expressed on a minor normal subset of B cells. CLL cells also express the B-cell antigens CD19, CD20 (low level), and CD23. Immunophenotyping can also be used for prognosis: high-level expression of CD38 and ZAP-70 is associated with worse prognosis.
Cytogenetics—Chromosomal abnormalities in CLL have prognostic significance. Deletions of 11q and 17p are associated with significantly shorter survival. Deletion of 13q is associated with better prognosis.
Molecular genetics—As with all B-cell lymphomas, the cells of CLL have clonally rearranged immunoglobulin genes. CLL with hypermutated immunoglobulin gene regions (compared with the baseline unmutated sequences) has a better prognosis. Unmutated immunoglobulin genes are associated with worse prognosis.
Hairy cell leukemia is an uncommon form of non-Hodgkin lymphoma. This disease generally occurs in men with a median age at diagnosis of 50 years. The male to female ratio is approximately 4:1. The clinical manifestations are primarily the result of infiltration of the tumor cells into the bone marrow, liver, and spleen. A significant clinical finding on physical examination is the often massive splenomegaly. The liver is also enlarged, but to a much lesser degree than the spleen. Marrow failure is common in this disease, resulting in pancytopenia and its associated complications. Patients generally present with splenomegaly, leukopenia with a relative decrease in monocytes, and an inaspirable bone marrow.
Morphology— The diagnosis of hairy cell leukemia is supported by the identification of lymphocytes with bean-shaped nuclei and fairly abundant gray cytoplasm, giving the cells a somewhat monocytic appearance. Fine cytoplasmic projections that have a hair-like appearance on Wright–Giemsa-stained smears give this entity its name.
Cytochemistry—The cells in hairy cell leukemia stain positively for acid phosphatase that is partially or completely resistant to removal on the addition of tartrate. This is known as TRAP, for tartrate-resistant acid phosphatase. TRAP-positive lymphocytes with fine cytoplasmic projections are highly consistent with a diagnosis of hairy cell leukemia.
Immunophenotyping—The hairy cells have a B-cell phenotype, with monoclonal surface immunoglobulin, CD19 (increased), and CD20 (increased). Antigens that are relatively specific for hairy cell leukemia include the interleukin 2 receptor alpha, CD25, as well as surface CD11c and CD103. Immunohistochemistry performed on bone marrow biopsies or spleen can be used to detect DBA44, which is relatively selective, although not specific, for hairy cell leukemia. These results are all consistent with the identification of hairy cell leukemia as a B-cell disorder.
Molecular genetics—The neoplastic B cells of hairy cell leukemia have clonally rearranged immunoglobulin genes. Recently most hairy cell leukemias were found to have a mutation of the BRAF gene (V600E) that was previously found in melanoma. This mutation is relatively specific for hairy cell leukemia and does not appear to affect disease prognosis.
The diagnosis of hairy cell leukemia is supported by the identification of lymphocytes with bean-shaped nuclei and fairly abundant gray cytoplasm, giving the cells a somewhat monocytic appearance. Fine cytoplasmic projections that have a hair-like appearance on Wright–Giemsa-stained smears give this entity its name.
The plasma cell dyscrasias are disorders in which there is an expansion of a single clone of immunoglobulin-secreting cells. This results in the appearance of high levels of complete or incomplete immunoglobulin molecules in the serum or urine. The monoclonal immunoglobulin in the serum is known as an M-component because it is found in the prototype disorder in this group of diseases, multiple myeloma. Incomplete immunoglobulins containing only light chains or only heavy chains may be produced in certain plasma cell dyscrasias. The free light chains, which are known as Bence-Jones proteins, may be excreted into the urine. The 5 disorders included in this grouping of plasma cell dyscrasias are plasma cell myeloma, Waldenström macroglobulinemia, heavy chain disease, primary amyloidosis, and monoclonal gammopathy of unknown significance (MGUS). Amyloidosis is discussed in Chapter 3, and the other entities are described as follows.
Plasma cell myeloma, also known as multiple myeloma, is a disorder resulting from proliferation of a single plasma cell clone that produces a monoclonal immunoglobulin. The median age for presentation is 62 years. The most frequent presenting symptom is bone pain resulting from osteolytic lesions produced by clusters of plasma cells infiltrating the bone. The bones most often affected are the skull, the ribs, the vertebrae, and the long bones of the extremities. Because patients with multiple myeloma are often anemic, fatigue and weakness are common presenting symptoms. Patients may also experience recurrent bacterial infections as a result of the leukopenia that occurs later in the disease. In addition, the passage of free light chains into the urine may result in “myeloma kidney” and lead to renal failure. The diagnosis of myeloma depends on the presence of a monoclonal protein in the serum or urine, and then the type of myeloma is further classified based on the severity of the disease (Table 13–2). A skeletal survey is included in the initial workup of myeloma to assess the extent of bone involvement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


