http://evolve.elsevier.com/Edmunds/NP/
 Top 100 drug.
Top 100 drug.
INDICATIONS
See also Table 37-1.
The drugs in this section generally are initiated by a rheumatologist. Patients on these medications are seen in the primary care setting. All patients with suspected rheumatoid arthritis (RA) should be referred to a specialist for evaluation and initiation of medication. Primary care providers should work with the specialist in monitoring the course of the disease and identifying possible adverse reactions. Therefore, this chapter emphasizes the principles of drug use and ways to monitor the effects of medications. Methotrexate is discussed in detail because it is by far the most commonly used disease-modifying antirheumatic drug (DMARD). It is also well tolerated and produces beneficial effects within 2 to 6 weeks. A new drug class on the market, JAK inhibitors, are to treat patients with moderate to severe rheumatoid arthritis particularly in adolescents.
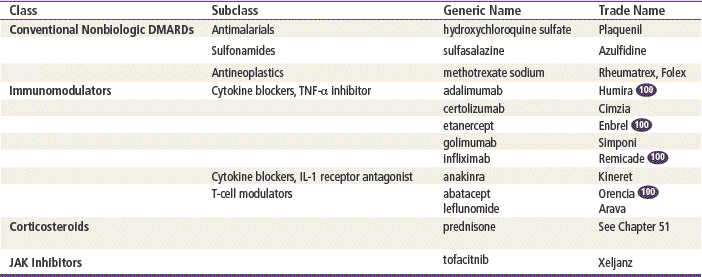
Drug treatment for RA continues to evolve. The conventional DMARDs methotrexate, hydroxychloroquine, and sulfasalazine remain in common use. Previously, gold salts, penicillamine, and azathioprine were used, but their use now is indicated only if all other therapy has failed because of the toxicity of these drugs. The newer immunomodulators are becoming increasingly important in the treatment of RA. The field continues to change rapidly as agents with new mechanisms of action are being developed. Many agents are currently being investigated in clinical trials.
Therapeutic Overview
Pathophysiology
The pathophysiology of RA is much the same as that of any autoimmune disease. For unknown reasons, the body fails to distinguish between self- and non–self-protein found in the body and proteins carried by foreign invaders. Current research suggests modest exposure to sulfur dioxide in air pollution may increase the risk of developing RA. In all individuals, it is not uncommon for a T– or B–immune cell lymphocyte to react to a self-protein during its development in the thymus or bone marrow. Normally, these self-reactive immune cells are destroyed, but occasionally, they escape destruction. Years later, they are activated to trigger an immune response. Activation is thought to occur after infection with a common bacterium or virus that contains a protein with a stretch of amino acids that match a stretch of amino acids on the tissue protein. The organisms most commonly implicated in this activation include Streptococcus, Mycoplasma, and Borrelia (the agent of Lyme disease), although retroviruses also may be responsible.
In RA, the causative agents first gain access to the joint and cause an inflammatory response. This results in damage to small blood vessels and leads to the accumulation of inflammatory cells (i.e., macrophages and lymphocytes). Macrophages process the pathogenic material and transfer the antigen to lymphocytes. Among the lymphocytes, the B-cells produce antibodies, and the T-cells produce cytokines that activate B-cells and cytotoxins that attack tissues directly inside the joint capsule, causing synovitis. As the disease progresses, the inflammatory process spreads from the synovium into the cartilage and bone, with collagen-destroying enzymes causing destruction of the joint. These changes begin within the first 2 years of the disease, making early diagnosis and aggressive treatment very important.
Disease Process
The clinical presentation of RA is extremely variable, but it is chiefly characterized by disfigurement and inflammation of multiple peripheral joints. Articular signs and symptoms include symmetric joint swelling with stiffness, warmth, tenderness, and pain. Stiffness is usually worse in the morning. Duration of stiffness is a measure that can be used to evaluate disease activity. Although any joint may be affected, the joints most often affected are the proximal interphalangeal and metacarpophalangeal joints and the wrists, knees, ankles, and toes. Systemic symptoms include a prodrome of malaise, fatigue, fever, and weight loss.
Physical examination reveals acute inflammation of the joint, seen as tenderness and swelling. Heat and redness are not prominent features of RA, although an involved joint is often warmer on examination. Anemia, high-spiking fever, rash, and other extraarticular features occur in systemic onset juvenile arthritis. Later in the disease, the examination becomes specific for RA. X-rays of the wrists or feet are usually the earliest way to detect changes but are not diagnostic in the early phase of RA. The erythrocyte sedimentation rate is a very nonspecific but sensitive indication of inflammation. It is elevated in active disease and usually is monitored as an indicator of the effectiveness of therapy. RA factor (an immunoglobulin [Ig]M antibody) is positive in 75% to 85% of patients with RA. Antinuclear antibodies are elevated in approximately 20% of patients with RA.
Over time, the disease involves skin and blood vessels, lymph tissues, eyes, chest cavity, lungs, nerves, and blood. Patients with RA have been shown in some studies to have an almost 40% higher risk of atrial fibrillation and a more than 30% greater risk of stroke than those in the general population. Patients with classic RA and the need for aggressive treatment have joint symptoms that persist beyond 2 years, positive rheumatoid factor, poor functional status, a large number of inflamed joints, and extraarticular manifestations of disease.
Mechanism of Action
DMARDs have antiinflammatory effects that may slow disease progression and preserve joint function. The exact mechanism of how they work in RA is unclear.
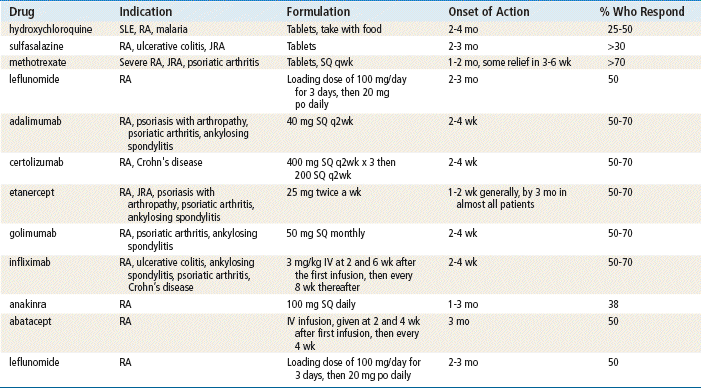
Hydroxychloroquine
It has been suggested that hydroxychloroquine sulfate acts by mimicking the action of its parent compound, chloroquine. The antimalarials possess antiinflammatory properties, which possibly are seen as inhibiting the conversion of arachidonic acid to prostaglandin F2. In vitro, these agents interfere with the chemotaxis of polymorphonuclear leukocytes, macrophages, and eosinophils.
Sulfonamides
The mechanism of action of sulfasalazine remains unknown. Sulfasalazine has antiinflammatory actions and reaches high concentrations in serous fluids in connective tissue.
Methotrexate
Methotrexate (MTX) is used because it possesses both antiinflammatory and immunosuppressive properties. Evidence of clinical efficacy has been shown in numerous studies. This agent acts by inhibiting dihydrofolate reductase and 5-amino-imidazole-4-carboxamide ribonucleotide transferamylase, resulting in impaired DNA synthesis. Furthermore, it exhibits inhibitory effects on cytokines, especially interleukin-1, and may alter arachidonic acid metabolism. Methotrexate also exerts an antiproliferative effect on synovial cells.
Immunomodulators
Agents that regulate the extent and duration of the immune response are known as immunomodulators. Cytokines such as tissue necrosis factor (TNF) and interleukin (IL) are some of the many substances the body releases during the inflammatory response. Through genetic research, these agents have been cloned so that they can be produced outside the body and given to patients who are immunodeficient. Many are indicated for the treatment of RA. Some are indicated for multiple sclerosis.
Cytokine Blockers
Cytokines are cellular messengers that initiate and perpetuate the inflammatory response by stimulating the production of cartilage-degrading enzymes and allowing pannus, an inflammatory exudate, to attach to cartilage and bone. The two cytokines targeted by the new immune modulators are TNF and IL-1.
The cytokine blockers–TNF inhibitors adalimumab, etanercept, and infliximab bind to TNF and inhibit its interaction with cell surface receptors. The cytokine blocker–IL-1, anakinra, blocks IL-1 by competitively inhibiting binding to the receptor. IL-1 mediates inflammatory responses and causes cartilage degradation. Abatacept is a T-cell modulator that inhibits T-cells by binding to CD80 and 86, thereby blocking interaction with CD28. Leflunomide blocks the synthesis of pyrimidine, which is required for proliferation of T-cells.
Corticosteroids
Corticosteroids inhibit the production of interleukins. This inhibition decreases the immune response through diminished activity of T-cells. Corticosteroids also affect both the type and the number of leukocytes and monocytes in serum. The antiinflammatory effects of corticosteroids overlap with their immunosuppressive actions. They affect carbohydrate, protein, and lipid metabolism and increase hepatic gluconeogenesis. Detailed information regarding corticosteroids can be found in Chapter 51.
Treatment Principles
Evidence-Based Recommendations
The DMARD methotrexate is widely used as first-line treatment in individuals with RA because of consensus about its effectiveness in practice.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


