INTRODUCTION
Through the ages, physicians and scientists have studied diseases of humans, describing the abnormal lesions (pathology) and their adverse effects on the patient (pathophysiology), seeking to identify risk factors (susceptibility) and causes (etiological agents), and pursuing a greater understanding of how diseases come to be (pathogenesis). With progress in these directions, some human diseases are now well understood from the perspective of risk and causal factors, and effective preventative and/or treatment strategies have been developed. For many other human diseases, investigation into the fundamental elements of the molecular and cellular pathogenesis represents an ongoing endeavor. Nevertheless, in the last several decades, the field of pathology has evolved to recognize that disease manifests in many cases as a direct reflection of changes in patterns of gene expression and often involves changes in the genome. There is also the recognition that gene expression patterns in a given lesion type (e.g., a certain form of cancer) will influence the clinical behavior of that lesion and its response to therapy. Hence, a great effort has been expended to characterize the genetic basis of various human diseases (molecular pathology), leading to a greater understanding of the contributions of genomic alterations to the development and progression of disease. Molecular pathology represents the application of the principles of basic molecular biology to the investigation of human disease processes. Our ever broadening insights into the molecular basis of disease provide opportunities for the development of new and novel approaches for diagnosis, classification, and prognostic assessment of human disease, and for expansion of treatments that are directed at specific molecular targets or pathways. In this chapter, we provide an overview of the role of the genomic alterations in genetic, developmental, and neoplastic diseases, with special emphasis on the role of mutations and epimutations in the pathogenesis of these disease states.
THE HUMAN GENOME
Among the essential building blocks of living cells that were identified and characterized by chemists and early biochemists were nucleic acids—long-chain polymers composed of nucleotides. Nucleic acids were named based partly on their chemical properties and partly on the observation that they represent a major constituent of the cell nucleus. The critical realization that nucleic acids form the chemical basis for the transmission of genetic traits did not occur until about 65 years ago. Prior to that time, there was considerable disagreement among scientists as to whether genetic information was contained in and transmitted by proteins or nucleic acids. It was recognized that chromosomes contained deoxyribonucleic acid (DNA) as a primary constituent, but it was not known if this DNA carried genetic information or merely served as a scaffold for some undiscovered class of proteins that carried genetic information. However, the demonstration that genetic traits could be transmitted through DNA formed the basis for numerous investigations focused on elucidation of the nature of the genetic code. During the last 65 years, numerous investigators participated in the scientific revolution leading to modern molecular biology. Of particular significance were the elucidation of the structure of DNA, determination of structure-function relationships between DNA and RNA, and acquisition of basic insights into the processes of DNA replication, RNA transcription, and protein synthesis.
Molecular biology has developed into a broad field of scientific pursuit and, at the same time, has come to represent a basic component of most other basic research sciences. This has come about through the rapid expansion of our insights into numerous basic aspects of molecular biology and the development of an understanding of the fundamental interaction among the several major processes that comprise the larger field of investigation. A theory, referred to as the central dogma, describes the interrelationships among these major processes. The central dogma defines the paradigm of molecular biology that genetic information is perpetuated as sequences of nucleic acid and that genes function through their expression in the form of protein molecules. Individual DNA molecules serve as templates for either complementary DNA strands during the process of replication or complementary RNA molecules during the process of transcription. In turn, RNA molecules serve as blueprints for the ordering of amino acids by ribosomes during protein synthesis or translation. This simple representation of the complex interactions and interrelationships among DNA, RNA, and protein (Figure 1-1A) was proposed and commonly accepted shortly after the discovery of the structure of DNA. Nonetheless, this paradigm still holds more than 50 years later and continues to represent a guiding principle for molecular biologists involved in all areas of basic biological, biomedical, and genetic research.
FIGURE 1-1
The central dogma. (A) The central dogma of molecular biology as originally described by Crick. This schematic illustrates the flow of genetic information from DNA to RNA and then to protein, as well as illustrates that DNA serves as its own template in replication. (B) The new central dogma reflects advances in our understanding of molecular processes that occur in normal cells. These processes include epigenetic regulation of gene expression by DNA methylation and histone modification, post-transcriptional regulation of gene expression by microRNAs, and modification of protein functionality by post-translational modification (which might include glycosylation, ubiquitination, phosphorylation, or other). This new central dogma also emphasizes the importance of DNA repair processes in the maintenance of genome integrity.

With advances in our understanding of the molecular underpinnings of the eukaryotic cell, the concept of the central dogma remains largely the same, but we now recognize the importance of maintenance of genomic integrity through DNA repair mechanisms, multiple mechanisms and layers of regulation of gene expression (at the transcriptional and post-transcriptional levels), and modification of protein functionality through post-translational modification (Figure 1-1B). Numerous DNA repair mechanisms are expressed in eukaryotic cells to ensure the integrity of the primary structure of the DNA (the DNA sequence). These mechanisms function in conjunction with the processes of replication (to protect against replication errors) and transcription (to protect expressed regions of the genome). Major DNA repair pathways for damage to the primary sequence of the DNA include mismatch repair, nucleotide excision repair, and direct chemical reversal mechanisms. Eukaryotic cells also have DNA repair pathways that can correct strand breaks and other damage at the chromosomal level. Transcriptional regulation of gene expression is accomplished through balanced interactions of specific activating transcription factors and repressive gene expression inhibitors. In addition, transcriptional control of gene expression is heavily influenced by epigenetic factors, including methylation of the DNA sequence and remodeling of chromatin secondary to modification of histone proteins. Post-transcriptional regulation of gene expression is influenced by microRNAs (miRNAs) that function to direct the degradation of specific mRNAs. The proteins encoded by structural genes are often modified post-translationally through glycosylation, phosphorylation, acetylation, methylation, or through enzymatic cleavage, resulting in a polypeptide with new or modified functionality. This new concept of the central dogma more accurately describes the number and complexity of cellular functions that impinge on the genome and its expression (Figure 1-1B).
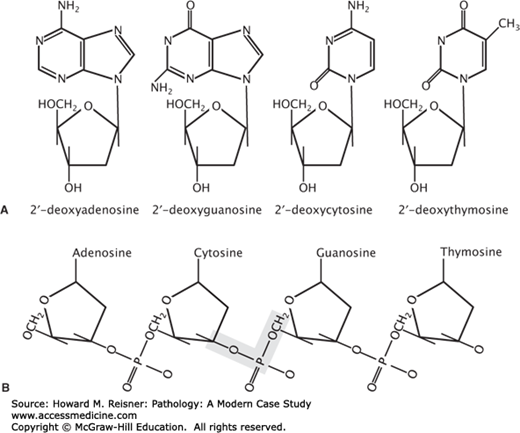
DNA is a polymeric molecule that is composed of repeating nucleotide subunits. The order of nucleotide subunits contained in the linear sequence or primary structure of these polymers represents all of the genetic information carried by a cell. Each nucleotide is composed of (i) a phosphate group, (ii) a pentose (5 carbons) sugar, and (iii) a cyclic nitrogen containing compound called a base. In DNA, the sugar moiety is 2-deoxyribose. Eukaryotic DNA is composed of four different bases: adenine, guanine, thymine, and cytosine (Figure 1-2A). These bases are classified based on their chemical structure into two groups: adenine and guanine are double-ring structures termed purines and thymine and cytosine are single-ring structures termed pyrimidines (Figure 1-2A). Within the overall composition of DNA, the concentration of thymine is always equal to the concentration of adenine, and the concentration of cytosine is always equal to guanine. Thus, the total concentration of pyrimidines always equals the total concentration of purines. The constant ratio of purines to pyrimidines in DNA, and more specifically the equal concentrations of adenine/thymine and guanosine/cytosine, is known as Chargaff rule. Chargaff observations related to the purine/pyrimidine composition of DNA was validated when it was recognized that adenine/thymine (A:T) and guanosine/cytosine (G:C) are specifically hydrogen bond in the structure of DNA. Adenine–thymine pairs are linked in the structure of DNA by double hydrogen bonding, and guanosine–cytosine pairs are linked by triple hydrogen bonding. The extensive hydrogen bonding between strands of DNA in the double helical structure of the molecule confers great stability under physiological conditions. The monomeric nucleotide units are linked together into the polymeric structure of DNA by 3′,5′-phosphodiester bonds (Figure 1-2B). Natural DNAs display widely varying sizes depending on the source. Relative molecular weights range from 1.6 × 106 daltons for bacteriophage DNA to 1 × 1011 daltons for a human chromosome.
FIGURE 1-2
Building blocks of DNA. (A) The chemical structure of purine (2′-deoxyadenosie and 2′-deoxyguanosine) and pyrimidine (2′-deoxycytosine and 2′-deoxythymosine) deoxyribonucleosides. (B) The chemical structure of repeating nucleotide subunits in DNA. The shaded area highlights a 3′-5′phosphodiester bond.
The structure of DNA is a double helix, composed of two polynucleotide strands that are coiled about one another in a spiral. Each polynucleotide strand is held together by phosphodiester bonds linking adjacent deoxyribose moieties (Figure 1-2B). The two polynucleotide strands are held together by a variety of noncovalent interactions, including lipophilic interactions between adjacent bases and hydrogen bonding between the bases on opposite strands. The sugar-phosphate backbones of the two complementary strands are antiparallel—that is, they possess opposite chemical polarity. Moving along the DNA double helix in one direction, the phosphodiester bonds in one strand are oriented 5′-3′, whereas in the complementary strand, the phosphodiester bonds are oriented 3′-5′. This configuration results in base pairs being stacked between the two chains perpendicular to the axis of the molecule. The base pairing is always specific: adenine is always paired to thymidine, and guanine is always paired to cytosine. This specificity results from the hydrogen-bonding capacities of the bases themselves. Adenine and thymine form two hydrogen bonds, and guanine and cytosine form three hydrogen bonds. The specificity of molecular interactions within the DNA molecule allows one to predict the sequence of nucleotides in one polynucleotide strand if the sequence of nucleotides in the complementary strand is known. Although the hydrogen bonds themselves are relatively weak, the number of hydrogen bonds within a DNA molecule results in a very stable molecule that does not spontaneously separate under physiological conditions. There are many possibilities of hydrogen bonding between pairs of heterocyclic bases. Most important are the hydrogen bonded base pairs A:T and G:C that were proposed by Watson and Crick in their double-helix structure of DNA. However, other forms of base pairing have been described. In addition, hydrophobic interactions between the stacked bases in the double helix lend additional stability to the DNA molecule. Three helical forms of DNA are recognized to exist: A, B, and Z. The B conformation is the dominant form under physiological conditions. In B DNA, the base pairs are stacked 0.34 nm apart, with 10 base pairs per turn of the right-handed double helix and a diameter of approximately 2 nm. Like B DNA, the A conformer is also a right-handed helix. However, A DNA exhibits a larger diameter (2.6 nm), with 11 bases per turn of the helix, and the bases are stacked closer together in the helix (0.25 nm apart). Careful examination of space-filling models of A and B DNA conformers reveals the presence of a major groove and a minor groove. These grooves (particularly the minor groove) contain water molecules that interact favorably with the amino and keto groups of the bases. In these grooves, DNA-binding proteins can interact with specific DNA sequences without disrupting the base pairing of the molecule. In contrast to the A and B conformers of DNA, Z DNA is a left-handed helix. Z DNA possesses a minor groove but no major groove, and the minor groove is sufficiently deep that it reaches the axis of the DNA helix. The natural occurrence and potential physiological significance of Z DNA in living cells has been the subject of much speculation, but is now gaining recognition. Additional non-B DNA structures have been described, some of which may contribute to the genetic basis of human disease.
The diploid genome of the typical human cell contains approximately 3 × 109 base pairs of DNA that is subdivided into 23 pairs of chromosomes (22 autosomes and sex chromosomes X and Y). For many years, it was suggested that discernment of the complete sequence of the human genome would enable the genetic causes of and contributions to human disease to be investigated. However, early molecular biology techniques and approaches to DNA cloning and sequencing were slow and cumbersome. Nevertheless, practical methods for DNA sequencing appeared in the mid to late 1970s, and numerous reports of DNA sequences corresponding to segments of the human genome began to appear. In the mid-1980s, a project to sequence the complete human genome was proposed, and this project began in the later years of that decade. The development of automated methods for DNA sequencing made the ambitious goals of the Human Genome Project attainable. Subsequently, detailed genetic and physical maps of the human genome appeared, expressed sequences were identified and characterized, and gene maps of the human genome were constructed. Efforts by several consortiums using differing approaches to large-scale sequencing of human DNA and sequence contig assembly culminated in 2001 with the publication of a draft sequence of the human genome. Several years later, a more complete sequence of the human genome was released. Today, the human genome is thought to contain approximately 21,000 distinct protein-coding genes. Analysis of the human genome sequence reveals considerable variability between individuals, including in excess of 1.1–1.4 million single-nucleotide polymorphisms (SNPs) distributed throughout the genome. Refinement of the sequence of the human genome, identification and characterization of the genes contained, and description of the features of the genome (SNPs and other variations) continue at a rapid pace (www.genome.gov). The implications for knowing the sequence of the human genome in the context of understanding the impact of genetic factors on human disease are enormous.
Human genomic DNA is packaged into discreet structural units that vary in size and genetic composition. The structural unit of DNA is the chromosome, which is a large continuous segment of DNA. A chromosome represents a single genetically specific DNA molecule to which a large number of protein molecules are attached that are involved in the maintenance of chromosome structure and regulation of gene expression. Genomic DNA contains both coding and noncoding sequences. Noncoding sequences contain information that does not lead to the synthesis of an active RNA molecule or protein. This is not to suggest that noncoding DNA serves no function within the genome. On the contrary, noncoding DNA sequences have been suggested to function in DNA packaging, chromosome structure, chromatin organization within the nucleus, and/or in the regulation of gene expression. A portion of the noncoding sequences represent intervening sequences that split the coding regions of structural genes. However, the majority of noncoding DNA falls into several families of repetitive DNA whose exact functions have not been entirely elucidated. Coding DNA sequences give rise to all of the transcribed RNAs of the cell, including mRNAs that encode for protein products. The organization of transcribed structural genes consists of coding regions that are interrupted by intervening noncoding regions of DNA. Thus, primary RNA transcripts contain both coding and noncoding sequences, and the noncoding sequences must be removed from the primary RNA transcript during processing to produce a functional mRNA molecule appropriate for translation.
DNA serves two essential functions with respect to cellular homeostasis: (i) storage of genetic information and (ii) transmission of genetic information. The DNA molecule serves as a template to fulfill each of these general functions. During cell division, DNA serves as a template for the faithful replication of genetic information that is ultimately passed into daughter cells. Likewise, the DNA molecule serves as a template for restoration of normal DNA sequence during DNA repair processes. During normal cellular operations, DNA serves as a template for the transcription of RNA. Transcribed RNA molecules may function directly (as is the case for ribosomal RNAs and transfer RNAs), may function after processing (as is the case of miRNAs), or may function as the template for synthesis of cellular proteins (as in the case of mRNAs).
Discovery of the double-stranded structure of DNA led rapidly to the suggestion that DNA replication could be accomplished in a semiconservative manner. In semiconservative DNA replication, each strand of the DNA helix serves as a template for the synthesis of the complementary strand. The result is the formation of two complete copies of the DNA molecule, each consisting of one strand from the parent DNA molecule and one newly synthesized strand. Utilization of the DNA strands as templates for the synthesis of complimentary DNA strands ensures the faithful reproduction of the genetic material for transmission into daughter cells.
Contained within the linear nucleotide sequence of DNA is the information necessary for the synthesis of all protein constituents of a cell, as well as numerous species of functional RNA molecules. Transcription is the process by which RNA molecules are synthesized with a sequence complimentary to the transcriptional unit of DNA. Hence, the mRNA that encodes a specific protein is complimentary to the DNA sequence of that specific gene. In similar fashion, rRNAs, tRNAs, and various other RNA species are transcribed. The correct start and end points for transcription of a particular gene are determined by the promoter sequence upstream of the coding sequence of the gene and by termination signals downstream. Various other regulatory elements are present in the human genome in association with transcribed sequences. Some of these regulatory elements are enhancers of transcription, while others provide for negative regulation (repression) of gene expression in response to specific physiological signals. In contrast to DNA replication where both strands of the molecule serve as templates for synthesis of complementary DNA strands, in RNA transcription, only one strand of the DNA serves as template. This strand is referred to as the sense strand. Transcription of the sense strand ultimately yields an mRNA molecule that encodes the proper amino acid sequence for a specific protein.
Genetic recombination represents one mechanism for the generation of genetic diversity through the exchange of genetic material between two homologous nucleotide sequences. Such an exchange of genetic material often results in alterations of the primary structure (nucleotide sequence) of a gene and alteration of the primary structure (amino acid sequence) of the encoded protein product. In organisms that reproduce sexually, recombination is initiated by formation of a junction between similar nucleotide sequences carried on the same chromosome from two different parents. The junction is able to move along the DNA helix through branch migration resulting in an exchange of the DNA strands (sister chromatid exchange).
The DNA contained in each of the cells of the human body is assaulted on a daily basis by various endogenous and exogenous DNA damaging agents, and some of this damage leads to mutation. DNA damage can result from spontaneous alteration of the DNA molecule or from the interaction of numerous chemical and physical agents with the structural DNA molecule. Spontaneous lesions can occur during normal cellular processes, such as DNA replication, DNA repair, or gene rearrangement, or through chemical alteration of the DNA molecule itself as a result of hydrolysis, oxidation, or methylation. In most cases, DNA lesions create nucleotide mismatches that lead to point mutations. Nucleotide mismatches can result from the formation of apurinic or apyrimidinic sites following depurination or depyrimidination reactions, nucleotide conversions involving deamination reactions, or in rare instances, from the presence of a tautomeric form of an individual nucleotide in replicating DNA. Deamination reactions result in the conversion of cytosine to uracil, adenine to hypoxanthine, and guanine to xanthine. However, the most common nucleotide deamination reaction involves methylated cytosines, which can replace cytosine in the linear sequence of a DNA molecule in the form of 5-methylcytosine. The 5-methylcytosine residues are always located next to guanine residues on the same chain, a motif referred to as a CpG dinucleotide. The deamination of 5-methylcytosine results in the formation of thymine. This particular deamination reaction accounts for a large percentage of spontaneous mutations in human disease. Interaction of DNA with physical agents, such as ionizing radiation, can lead to single-strand or double-strand breaks as a result of scission of phosphodiester bonds on one or both polynucleotide strands of the DNA molecule. Ultraviolet (UV) light can produce different forms of photoproducts, including pyrimidine dimers between adjacent pyrimidine bases on the same DNA strand. Other minor forms of DNA damage caused by UV light include strand breaks and cross-links. Nucleotide base modifications can result from exposure of the DNA to various chemical agents, including N-nitroso compounds and polycyclic aromatic hydrocarbons. DNA damage can also be caused by chemicals that intercalate the DNA molecule and/or cross-link the DNA strands. Bifunctional alkylating agents can cause both intrastrand and interstrand cross-links in the DNA molecule.
Mutation is simply defined as any permanent change to the DNA molecule. The various forms of spontaneous and induced DNA damage give rise to a plethora of different types of molecular alterations to the DNA molecule, leading to stable mutations. These various types of mutation include both gross alteration of chromosomes and more subtle alterations to specific gene sequences in otherwise normal chromosomes.
Gross chromosomal aberrations include large deletions, additions (reflecting amplification of DNA sequences), translocations (reciprocal and nonreciprocal), and other forms of chromosomal rearrangement. All of these forms of chromosomal abnormality can be distinguished through standard karyotype analyses of G-banded or R-banded chromosomes. However, currently employed chromosomal analysis methodologies typically rely on fluorescence in situ hybridization (FISH) and derived techniques. FISH can be utilized to address questions related to a single genetic locus or region (to score for amplification or deletion), or might be applied more broadly as a means for karyotypic analysis. Spectral karyotyping (or SKY) is a method where FISH probes paint individual chromosomes a distinct color, enabling easy and straightforward enumeration of chromosome numbers and identification of structural rearrangements.
The major consequence of chromosomal deletion is the loss of specific genes that are located in the deleted chromosomal region, resulting in changes in the copy number of the affected genes. For instance, deletion of certain classes of genes, including tumor suppressor genes or genes encoding the proteins involved in DNA repair, leads to predisposition of affected cells to neoplastic transformation. Likewise, amplification of chromosomal regions results in an increase in gene copy numbers, which can lead to the same circumstance if the affected region contains genes for dominant proto-oncogenes or other positive mediators of cell cycle progression and proliferation. The direct result of chromosomal translocation is the movement of some segment of DNA from its natural location into a new location within the genome, which can result in altered expression of the genes that are contained within the translocated region. If the chromosomal breakpoints utilized in a translocation are located within structural genes, then new hybrid genes can be generated. Likewise, some chromosomal rearrangements cause loss of structural genes due to the presence of a breakpoint within the gene itself.
The most common forms of mutation involve single-nucleotide alterations, small deletions, or small insertions into specific gene sequences. Unlike chromosomal alterations, these molecular changes to the genome can only be detected through DNA sequencing or other sensitive molecular analyses that employ sequence-specific PCR primers.
Single-nucleotide alterations that involve a change in the normal coding sequence of the gene are referred to as point mutations. The consequence of most point mutations is an alteration in the amino acid sequence of the encoded protein. However, some point mutations are silent and do not affect the structure of the gene product. Silent mutations (also known as synonymous mutations) are possible because most amino acids are encoded by more than one triplet codon. Silent mutations represent one mechanism for introduction of genetic variability, but are without functional consequence. The remaining point mutations fall into two classes: (i) missense mutations and (ii) nonsense mutations. Missense mutations involve nucleotide base substitutions that alter the translation of the affected codon triplet, very often with a nonconservative amino acid change. Missense mutations are frequently classified as transitions or transversions based on the nature of the nucleotide change in the sequence of the DNA. A transition is the replacement of a nucleotide by another nucleotide of the same chemical class (i.e., purine for purine or pyrimidine for pyrimidine). Transition point mutations include A to G, G to A, C to T, and T to C. In contrast, a transversion is the substitution of a nucleotide from one chemical class with one from the other chemical class (i.e., purine for pyrimidine or pyrimidine for purine). Transversion point mutations include A to T, A to C, G to C, G to T, T to A, T to G, C to A, and C to G. Nonsense mutations differ from missense mutations in that they involve nucleotide base substitutions that modify a triplet codon that normally encodes for an amino acid into a translational stop codon. This results in the premature termination of translation and the production of a truncated protein product. In some cases, the truncated protein product resulting from a missense mutation lacks functionality and is rapidly degraded. In other cases, the truncated protein may retain some function or may gain new functionality (like freedom from normal regulatory mechanisms).
Small deletions and insertions usually give rise to frameshift mutations because the deletion or insertion of a single nucleotide (for instance) alters the reading frame of the gene on the 3′ side of the affected site. This results in the synthesis of a polypeptide product that does not resemble the normal gene product. In addition, small insertions or deletions can result in the premature termination of translation resulting from the presence of a stop codon in the new reading frame of the mutated gene. Deletions or insertions that occur involving multiples of three nucleotides will not result in a frameshift mutation, but will alter the resulting polypeptide gene product, which will exhibit either loss of specific amino acids or the presence of additional amino acids within its primary structure. These types of alteration can also lead to a loss of protein function.
All cellular DNA is subject to mutation through spontaneous mechanisms or in response to mutagenic agents. When DNA damaging and mutational events affect the DNA of a somatic cell, the resulting mutations are referred to as somatic mutations. Somatic cells represent the cells that compose all of the tissues in the organism outside of the germ line. Somatic mutations have been implicated in numerous disease types, particularly those that are characterized by clonal proliferation of altered cells (like in the case of cancer). Some somatic mutations occur during development, resulting in specific kinds of localized developmental abnormalities. Somatic mutations are not heritable. In contrast, DNA damaging and mutational events affecting the DNA of cells in the germ line produce mutations that are referred to as germ-line mutations. This distinction is biologically and clinically significant. The cells of the germ line are responsible for the genesis of oocytes (in the female) and spermatocytes (in the male). Hence, transmission of a germ-line mutation introduces a nucleotide sequence alteration into the fertilized egg. If that egg produces a viable organism, the mutation will be found in every cell in the organism. Germ-line mutations account for numerous genetic diseases, which can manifest systemic or organ-system-specific diseases (single organ or multiorgan). In addition, the germ-line mutation is found in the germ line of the affected individual, meaning that their offspring might also inherit the mutation.
THE HUMAN EPIGENOME
Conrad Waddington coined the term epigenetics in 1942 and defined it as “…the causal interactions between genes and their products, which bring the phenotype into being…” Today epigenetics refers to heritable alterations in gene expression patterns that are due to mechanisms other than changes in the nucleotide sequence of the genomic DNA. Epigenetic alterations differ from genetic alterations in that they arise more frequently, are reversible, and occur at defined regions of specific genes. Epigenetics explains how the same genotype can produce different phenotypes, such as in the case of monozygotic twins. Epigenetic information is stored as chemical modifications to (i) the histone proteins that package the genome and (ii) cytosines within the sequence of the DNA. These chemical changes regulate DNA accessibility and directly influence how the genome is expressed throughout different developmental stages, across different tissue and cell types, and in various disease states. Many epigenomic modifications affect DNA, RNA, and protein, but DNA methylation, histone modifications, chromatin remodeling factors, and noncoding RNAs are among the most well-studied epigenetic mechanisms. Numerous epigenomic processes function together to establish and preserve the global or site-specific open or closed chromatin states that ultimately determine whether a gene is active or inactive.
DNA methylation is regarded as the hallmark of epigenetic modifications. This chemical modification on cytosine bases provides heritable epigenetic information that is transmitted during DNA replication. DNA methylation is found in various bacteriophages, bacteria (without chromatin), fungi, plants, and mammals. However, DNA methylation is not found in model organisms such as Caenorhabditis elegans (worms) and Saccharomyces cerevisiae (budding yeast). Most of these organisms appear to survive when their genomic DNA methylation is dramatically decreased, but when DNA methylation is completely lost in mice embryonic lethality during organ development ensues. DNA methylation is a well-studied epigenetic mechanism that contributes to genetic instability and to the silencing of specific genes.
DNA methylation occurs almost exclusively on cytosines within CpG dinucleotides, which are found in the genome at approximately 20% of the predicted frequency, and the majority (>70%) of CpG dinucleotides are methylated. However, regions of CpG density, termed CpG islands, occur in the promoter regions of numerous genes proximal to their transcription start site, and/or in sequences adjacent to the promoter region (like exon 1). It has been suggested that up to 50% of all human genes may contain a promoter CpG island (Figure 1-3). These CpG islands are conventionally defined as ≥200 base pairs with ≥50% G+C and ≥0.6 CpG observed/CpG expected, although a more rigorous definition (≥200 base pairs with ≥60% G+C and ≥0.7 CpG observed/CpG expected) has been proposed. Some studies have suggested that large CpG islands are not required for sensitivity of gene promoters to methylation-dependent silencing. Rather, other regions of CpG density can behave like classically defined CpG islands. Most promoter CpG islands are unmethylated in normal tissues, but may become methylated in various pathological conditions. Distinct patterns of DNA methylation are found in the genomes of cells under different physiological conditions and in development. During most developmental stages DNA is heavily methylated. However, promoters and other regulatory regions of housekeeping genes are largely unmethylated in most cell types, and tissue-specific expressed genes are unmethylated only in the cell types where these genes are transcriptionally active.



