Disease
OBJECTIVES
The reader will be able to:
- List at least four diseases in which the pharmacokinetics of drugs is known to be altered.
- List and briefly discuss the pharmacokinetic parameters that are often altered in patients with hepatic and renal diseases.
- Judge, using pharmacokinetic principles, on when alteration of drug administration should be considered for patients with hepatic or renal diseases.
- Estimate how much the clearance of a drug of known fraction excreted unchanged is decreased in a patient with known renal function.
- Estimate the creatinine clearance of a patient from the patient’s age, weight, gender, and serum creatinine.
- List the assumptions that underlie the application of renal function tests in dosage regimen adjustment for patients with renal disease.
- Establish, from pharmacokinetic principles, a dosage regimen for a drug with no active metabolites in a patient of a given age, weight, gender, and serum creatinine, when the fraction excreted unchanged is known.
- Sketch the amount of drug in the body with time following the dosage regimen recommended for the patient in the previous objective.
- Discuss the pertinence of issues, such as diminished metabolism and transport in multiple organs, and metabolism in the kidneys, on the adjustment of drug administration in renal disease.
- Define clinical dialyzability, continuous ambulatory peritoneal dialysis, dialysis, dialysis clearance, dialyzer, dialyzer efficiency, extracorporeal dialysis, and hemodialysis.
- Calculate the changes in clearance and half-life of a drug and predict the kinetic events brought about by hemodialysis or continuous ambulatory peritoneal dialysis, given the clearance by the procedure, together with the total (body) clearance and the half-life in the absence of the dialysis treatment.
- Given dialysis clearance and the pharmacokinetic parameters of a drug in the absence of the dialysis procedure, anticipate if a supplementary dose is desirable immediately after dialysis. Also, determine what the supplementary dose should be.
 isease is a major source of variability in drug response. For many diseases, this variability is primarily a result of differences in pharmacokinetics, the area of principal focus in this chapter. Needless to say, drug therapy in each disease state may need to be tailored to the severity of the disease and the specific needs of the individual being treated. One must also be cognizant of the presence of diseases other than the one for which a given drug is taken. Concurrent diseases such as hepatic cirrhosis, congestive cardiac failure, and renal function impairment can alter dosage requirements as well. It is in this context that the effect of disease on drug therapy is examined in this chapter. Hepatic, cardiovascular, and renal disease are of primary importance, especially renal disease for which adjustment in dosage is the most quantitative.
isease is a major source of variability in drug response. For many diseases, this variability is primarily a result of differences in pharmacokinetics, the area of principal focus in this chapter. Needless to say, drug therapy in each disease state may need to be tailored to the severity of the disease and the specific needs of the individual being treated. One must also be cognizant of the presence of diseases other than the one for which a given drug is taken. Concurrent diseases such as hepatic cirrhosis, congestive cardiac failure, and renal function impairment can alter dosage requirements as well. It is in this context that the effect of disease on drug therapy is examined in this chapter. Hepatic, cardiovascular, and renal disease are of primary importance, especially renal disease for which adjustment in dosage is the most quantitative.
Hemodialysis and related procedures have become established treatments for patients with end-stage renal disease. These procedures are designed to remove toxic waste products that accumulate in patients with this disease. However, they also remove drugs. Thus, such procedures may require adjustment of drug administration, in addition to that required by the loss of renal function. This chapter provides information needed to decide when and how to make such an adjustment. Continuous ambulatory peritoneal dialysis is also discussed, because it is regarded as the treatment of choice for some patients. The chapter ends with a discussion of hemodialysis of overdosed patients to help remove drugs or other substances from the body.
HEPATIC DISEASES
Because the liver is the major site for drug metabolism, an impression prevails that special care should be taken when administering drugs to patients with disease states in which hepatic function is modified. Objective data, although generally supporting this impression, are occasionally in conflict.
One reason for the conflict arises from an attempt to classify hepatic disorders as a single entity. However, disorders of the liver, local or diffuse, are caused by many diseases; each disease affects various levels of hepatic organization to a different extent. With few exceptions, the hepatic clearance of drugs is, on average, decreased in cirrhosis. In contrast, in acute viral hepatitis, there appears to be a fairly even division between those drugs for which metabolic clearance is decreased, or half-life prolonged, and those for which no significant change is detected. Existing data suggest that drug elimination is diminished in obstructive jaundice. The effect is expected to be more extensive for those drugs eliminated predominantly by biliary excretion.
Another potential pitfall is to equate prolongation of half-life with diminution of hepatic drug-metabolizing activity. Half-life is controlled by both total clearance and volume of distribution, two independent parameters (Chapter 5, Elimination). To assess clearance and volume of distribution, the drug should be given intravenously to ensure complete bioavailability. When so studied, the volumes of distribution of some drugs remain unaltered in hepatic disease, but those of others are increased. An increase in volume of distribution is found particularly with drugs bound to albumin in patients with cirrhosis. The explanation lies in the depressed synthesis of albumin in these patients. The resultant fall in albumin concentration is responsible for a decreased plasma binding, an associated increase in volume of distribution, and an associated increase in clearance for a drug of low extraction. A fall in hepatic enzymes is responsible, in large part, for a diminished hepatic unbound clearance of many drugs. Oxidized drugs appear to be more affected than those eliminated by conjugation.
The influence of hepatic disease on drug absorption is incompletely understood. The problem is complicated by the need to separate disposition from absorption when analyzing plasma concentration–time data. However, it is likely that the oral bioavailability of drugs that are usually highly extracted by the liver is increased in cirrhosis (Fig. 15-1). There are two reasons for this increase. One is a diminished first-pass hepatic loss resulting from depressed hepatocellular activity. The other is that many cirrhotic patients develop portal bypass, a condition in which a significant fraction of the portal blood bypasses the functional liver cells and enters the superior vena cava via esophageal varices. These portacaval shunts, formed as a consequence of the portal hypertension brought about by the diseased liver, and the changes in the liver itself, can greatly increase oral bioavailability.
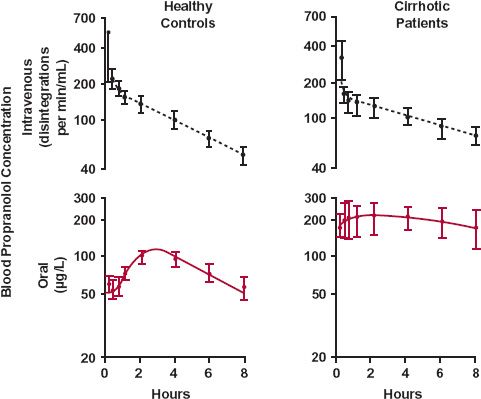
FIGURE 15-1. In cirrhosis, the oral bioavailability of propranolol is greatly increased, as evidenced by comparison of the blood concentration of unlabeled drug after oral administration (colored line) with that of tritiated drug after intravenous administration (dashed black line), following simultaneous determination of the kinetics of propranolol during the seventh dosing interval of an oral 8-hr dosing regimen in 9 normal subjects and 7 patients with cirrhosis (mean ± SE). (From: Wood AJJ, Kornhauser DM, Wilkinson GR, et al. The influence of cirrhosis on steady-state blood concentrations of unbound propranolol after oral administration. Clin Pharmacokinet 1978;3:478–487. Reproduced by permission of ADIS Press Australasia Pty Limited.)
From the foregoing discussion, it is apparent that drug dosage may need to be reduced in patients with hepatic function impairment as a result of both decreased clearance and increased oral bioavailability. An adjustment in dosage is particularly warranted when the usual regimen results in the unbound drug concentration at plateau approaching or exceeding the upper limit of the therapeutic concentration window. This condition arises when the clearance based on unbound drug is substantially depressed, because it is this clearance that controls the average unbound drug concentration at plateau, as 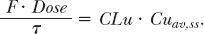
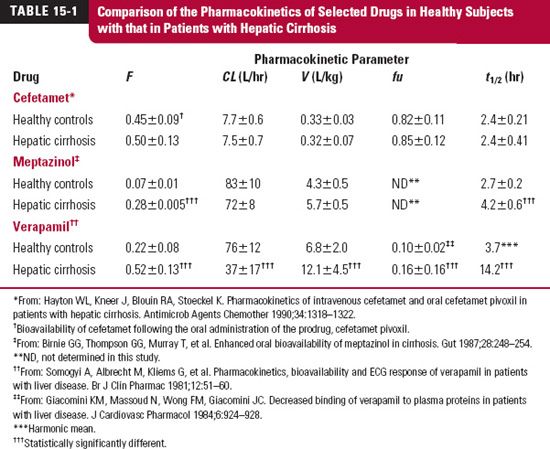
To illustrate these points, consider the data in Table 15-1. The observations on three different drugs studied in healthy subjects and in cirrhotic patients are shown. For cefetamet, a cephalosporin antibiotic, there is virtually no significant change in any of the pharmacokinetic parameters measured. This lack of an effect can be explained by this drug being primarily eliminated via renal excretion (fe = 0.85) and by its being only weakly bound to plasma proteins (fu = 0.85 in control subjects). Thus, changes in elimination and protein binding are not expected. On the other hand, meptazinol, an opioid analgesic, shows a dramatic increase in its bioavailability (400 + %) and about a 50% increase in half-life. This change resulting from hepatic cirrhosis, too, may be anticipated. One might expect meptazinol to have a high hepatic extraction ratio, for two reasons—the low oral bioavailability and the high plasma clearance. The increase in half-life is a result of a small, although not significant, change in both clearance and volume. With a high extraction ratio, the bioavailability reflects a decrease in hepatocellular activity and increased portacaval shunting of blood, common to hepatic cirrhosis. For verapamil, an antihypertensive and antiarrhythmic agent, significant changes occur in all of the pharmacokinetic parameters listed in the table. The drug has a high extraction ratio in controls, which decreases to an intermediate value in cirrhosis, as evidenced by the increase in F and the decrease in CL (determined from intravenous data). The volume of distribution based on the unbound drug (V/fu) is basically unchanged. The increase in half-life is then a consequence of a large decrease in unbound clearance (CL/fu).
Hepatic dysfunction is a graded phenomenon, and theoretically, a correlation should exist between changes in the pharmacokinetic parameters of drugs, especially hepatic clearance, and an appropriate measure of hepatic function. Attempts to establish such relationships, although encouraging, have not been highly successful. One example of an approach that has been partially successful is to relate clearance to the status of the hepatic function in hepatic cirrhosis.
In cirrhosis, a between correlation the severity of the condition (Child-Pugh Classification, Table 15-2) and the likelihood of depressed metabolism by oxidation, as shown for amprenavir (Agenerase, now also commercially available as a prodrug, fosamprenavir [Lexiva or Telzir]) in Fig. 15-2, and some conjugation reactions have been reported. Drug metabolism is often decreased in severe cirrhosis, signified by the combination of a low albumin (<28 g/L), an elevated clotting time (INR >2.2), refractory ascites, and the presence of Grade III or IV hepatic encephalopathies. The decreased metabolism frequently requires reducing the dose and monitoring the patient for adverse reactions.
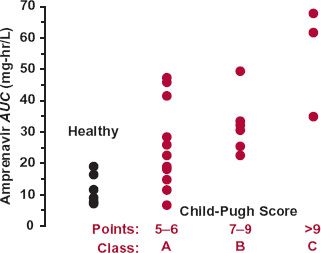
FIGURE 15-2. Relationship between amprenavir AUC and the Child-Pugh score. No subject with a Child-Pugh score greater than 12 was enrolled in the study. Amprenavir is eliminated primarily by CYP3A4 metabolism. Although there is considerable variability in patients with hepatic disease, there appears to be a trend toward an increased AUC, and therefore reduced clearance, in those with more severe hepatic disease. (From: Veronese L, Rautaureau J, Sadler BM, et al. Single-dose pharmacokinetics of amprenavir, a human immunodeficiency virus type 1 protease inhibitor, in subjects with normal or impaired hepatic function. Antimicrob Agents Chemother 2000;44:821–826.)
One needs to consider if an extensively metabolized drug is truly needed or if an alternative (renally excreted) drug is available.
One reason why the correlation with the Child-Pugh score often fails is because, unlike renal excretion, there are numerous pathways of drug metabolism, each with a different set of cofactor requirements and each affected to a different degree in hepatic disorders.
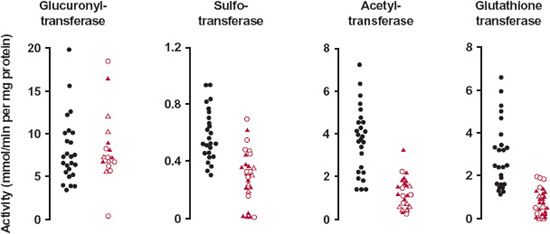
FIGURE 15-3. Activities of the conjugating enzymes, glucuronyltransferase, sulfotransferase, acetyltransferase, and glutathione transferase, in normal (black) and abnormal (colored) human livers vary widely. Open circles, filled triangles, and open triangles refer to biopsied samples from patients with chronic persistent hepatitis, chronic active hepatitis, and cirrhosis, respectively. The substrates were 2-naphthol for glucuronyl transferase and sulfotransferase, p-aminobenzoic acid for acetyltransferase, and benzo(a)pyrene-4,5-oxide for glutathione transferase. In contrast to the other enzymes, glucuronyl transferase does not appear to be affected by any of the hepatic conditions. Also apparent is a virtual lack of activity of the last three enzymes in some patients. The implications for drugs primarily eliminated by the conjugation pathways are great. (From: Pacifici GM, Viani A, Franchi M, et al. Conjugation pathways in liver disease. Br J Clin Pharmacol 1990;30:427–435.)
Variability in metabolism is apparent from the activities of four hepatic enzymes responsible for conjugation reactions (Fig. 15-3). The enzyme activities were measured in liver samples obtained by biopsy. Glucuronyl transferase appears to be unaffected by hepatic disease. Supporting this observation, the pharmacokinetics of the benzodiazepine oxazepam, which is eliminated almost entirely by glucuronidation, appears to be affected only in severe hepatic disease. It is also apparent that although the activities of the other three enzymes are on average decreased, there are some individuals within the normal range of activity and others in whom very little or no activity remains. Altered metabolism in chronic persistent hepatitis, chronic active hepatitis, and cirrhosis is also dependent on transporters, if involved.
CARDIOVASCULAR DISEASES
Circulatory disorders, which include shock, malignant hypertension, and congestive cardiac failure, are generally characterized by diminished vascular perfusion to one or more parts of the body. Because blood flow may influence drug absorption, distribution, and elimination, it is not surprising that the pharmacokinetics of drugs may be altered in circulatory disorders.
A diminished perfusion of absorption sites (e.g., gastrointestinal tract and muscle) with associated protracted and erratic drug absorption, tends to be seen in patients with depressed cardiovascular states; it may be necessary to give the drug intravenously if a prompt response is desired. However, in these conditions, the kinetics of distribution is also affected, with perfusion to many organs diminished. Exceptions are the brain and the myocardium, which consequently receive an increased fraction of an intravenous (i.v.) bolus dose, particularly in the earlier moments. For centrally acting and cardioactive agents, the rate of administration of a bolus dose to patients with circulatory depression must be tempered if the risk of toxicity is to be reduced. In these depressed circulatory states, cardiac output and therefore hepatic blood flow and, to a much lesser extent, renal blood flow are also reduced. Thus, a decrease in clearance of highly extracted drugs is expected. Evidence supporting this concept is illustrated in Fig. 15-4 by a strong positive correlation between the clearances of lidocaine, a drug eliminated hepatically by metabolism, and indocyanine green, a diagnostic agent rapidly picked up by active transporters within the liver, and excreted into bile, in patients with varying degrees of congestive cardiac failure. Both indocyanine green and lidocaine are high hepatic extraction ratio drugs whose clearances should therefore reflect a diminished hepatic blood flow. Notice in Fig. 15-4 the almost 16-fold variation in clearance of both lidocaine and indocyanine green. This range is almost certainly greater than the range of hepatic blood flows in these patients. The hepatic flow, usually about 18 mL/min per kilogram, is unlikely to fall to a value as low as 2 mL/min per kilogram, because severe anoxia is expected to result at even higher flow rates. Hepatocellular metabolism and transport are most probably also depressed when perfusion is severely diminished; this would further depress the clearances of both lidocaine and indocyanine green. Exposure to lidocaine is increased in patients with congestive cardiac failure, and the risk of toxicity is therefore increased.
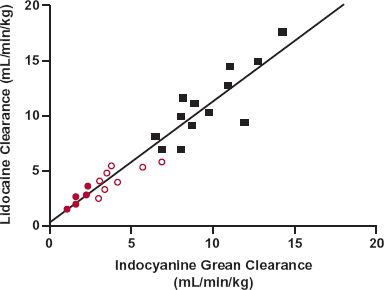
FIGURE 15-4. A strong positive correlation exists between the clearances of lidocaine and indocyanine green in patients without ( ) and with both mild (
) and with both mild ( ) and severe (
) and severe ( ) congestive cardiac failure. (From: Zito RA, Reid PR. Lidocaine kinetics predicted by indocyanine green clearance. Reprinted, by permission of New Engl J Med 1978;298:1160–1163.)
) congestive cardiac failure. (From: Zito RA, Reid PR. Lidocaine kinetics predicted by indocyanine green clearance. Reprinted, by permission of New Engl J Med 1978;298:1160–1163.)
RENAL DISEASES
There are many diseases of the kidneys. Some affect the glomerulus; others primarily affect the tubules. To a first approximation, the renal clearance of virtually all small molecules is reduced to the same degree regardless of the location of the disease. This general observation supports the “unit nephron hypothesis,” that is, there appears to be a corresponding decrease in filtration, secretion, and reabsorption. Stated differently, the decrease in renal clearance of a drug that is filtered and neither reabsorbed nor secreted is reduced in approximately the same proportion to one that is either highly secreted or extensively reabsorbed.
THE PHARMACOTHERAPEUTIC PROBLEM
In patients with compromised renal function, renal clearance of drugs is diminished. The degree of reduction depends on the reduction in renal function, as shown in Fig. 15-5 for cefepime, a fourth-generation cephalosporin antibiotic. The lower the renal function, the longer also is the half-life of the drug.
When a usual intramuscular (i.m.) regimen of amikacin (7.5 mg/kg) is administered to a 67-kg patient with renal function that is only 17% of a typical patient (colored line of Fig. 15-6), the drug accumulates extensively, relative to that in a typical patient (black line). Note also that the average amount in the body at steady state increases sixfold, whereas the peak amount only doubles. The trough value increases many fold. The figure reminds us that the extent of accumulation depends on both frequency of administration and half-life, and that the time required to approach plateau is a function of half-life only (Chapter 11, Multiple-Dose Regimens). Having a much longer half-life than usual, the time to reach steady state is much longer in this patient than in a individual with renal function of the typical patient. Obviously, to avoid excessive exposure, dosage must be reduced in the patient with renal dysfunction. The therapeutic questions to be asked here are whether the effects of the drug relate to the peak or average exposures to the drug. The former would suggest only a twofold reduction in dosage; the latter would require a sixfold decrease in dosage.
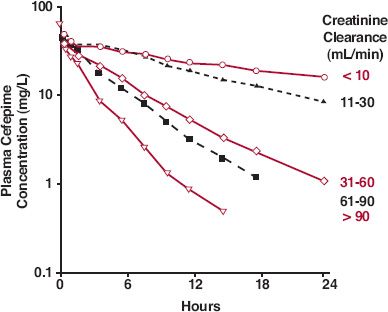
FIGURE 15-5. The mean plasma concentration–time profiles of cefepime, a cephalosporin antibiotic, are different in patients with varying degrees of renal function after i.v. infusion of a 1000-mg dose over 30 min. The subjects were grouped according to their measured creati-nine clearance values (in mL/min). (From: Barbhaiya RH, Knupp CA, Fargue ST, et al. Pharmacokinetics of cefepime in subjects with renal insufficiency. Clin Pharmacol Ther 1990;48:268–276.)
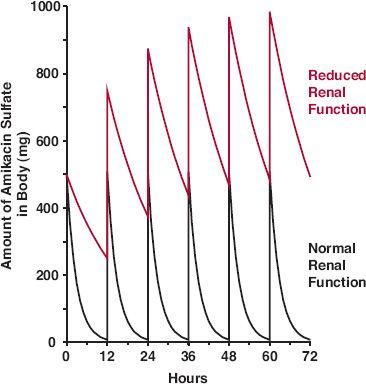
FIGURE 15-6. Sketch of the amount of amikacin sulfate in the body with time following a regimen of 500 mg every 12 hr in a patient whose renal function is normal (black line) and in a patient whose age and weight are the same but whose renal function is 17% of normal (colored line). Intravenous bolus administration is simulated. The normal half-life is assumed to be 2 hr.
The clinician needs information on which drugs result in excessive exposures in renal dysfunction and, more importantly, how to adjust drug administration to achieve an optimal therapeutic response. The basic principles that permit this calculation follow. These are developed with the view that drug effect, and therefore unbound drug concentration, needs to be maintained and that metabolites, if any, are inactive. The issue of active metabolites is further considered in Chapter 20, Metabolites and Drug Response.
DECREASE IN UNBOUND CLEARANCE
The elements of the problem of renal dysfunction are shown in Fig. 15-7. Ceftazidime clearance, unbound clearance here because the drug is not bound to plasma proteins, is low when renal function, measured by creatinine clearance, is low and increases linearly with renal function. Note that some unbound clearance remains (y-intercept) even when there is no renal function, as measured by creatinine clearance. This represents elimination by nonrenal pathways. The magnitude of change in unbound clearance depends on the renal function remaining and the fraction excreted unchanged.
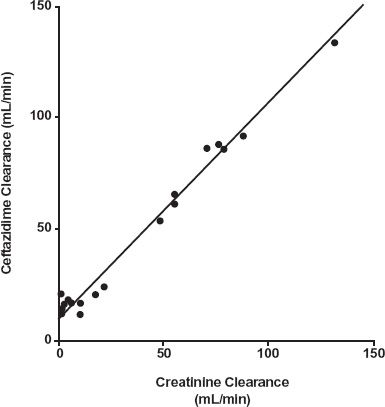
FIGURE 15-7. The total clearance of the cephalosporin, ceftazidime, varies linearly with creatinine clearance in a group of 19 patients with varying degrees of renal function. Note that some clearance remains (y-intercept) when there is no renal function. (From: van Dalen R, Vree TB, Baars AM, et al. Dosage adjustment for ceftazidime in patients with impaired renal function. Eur J Clin Pharmacol 1986;30:597–605.)
In what follows, emphasis is placed on unbound rather than total clearance. This is not only because the all-important unbound concentration is related to unbound clearance but also because, for many drugs, fu varies in renal disease, making interpretation based on total plasma concentration more problematic. Our goal is to devise a relationship that allows estimation of the maintenance regimen in a patient with renal impairment. The first step is to relate unbound renal clearance in the patient with renal disease (d), CLuR(d), to unbound renal clearance in the typical (t) 60-year-old, 70-kg patient, CLuR(t).
The ratio of the two is the renal function (RF) in the patient relative to that in the typical patient. This relative value is subsequently simply called renal function.
In the typical patient, CLuR(t) is a fraction, fe(t), of unbound (renal + nonrenal) clearance, CLu(t).
By combining Eqs. 15-1 and 15-2, the unbound renal clearance in the patient can be related to CLu(t), the unbound clearance (by all routes) in the typical patient, by
CLuR(d) = RF · fe(t) · CLu(t) 15-3
The next step is to consider the unbound clearance by nonrenal routes, CLuNR. In the typical patient, it is
CLuNR = [1 – fe(t)] · CLu(t) 15-4
CLuNR is usually not materially changed because of renal disease. This is not always the case as will be subsequently discussed. Its value is, however, expected to change with age and weight. On the assumption that it does so like renal function, then CLuNR(d)/CLuNR(t) is just the ratio of expected renal clearances with age and body weight. From Eq. 14-3 (Chapter 14, Age, Weight, and Gender) this ratio, the factor by which renal clearance deviates from that in the typical patient, is 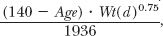 where the denominator is the value of the numerator when age is 60 years and weight is 70 kg. Based on these considerations, the factor by which nonrenal clearance (CLuNR[d]) deviates from that in the typical patient (CLuNR[t]) then becomes
where the denominator is the value of the numerator when age is 60 years and weight is 70 kg. Based on these considerations, the factor by which nonrenal clearance (CLuNR[d]) deviates from that in the typical patient (CLuNR[t]) then becomes
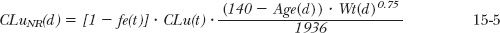
where age and weight are in years and kilograms, respectively. Now, under any condition,
CLu(d) = CLuR(d) + CLuNR(d) 15-6
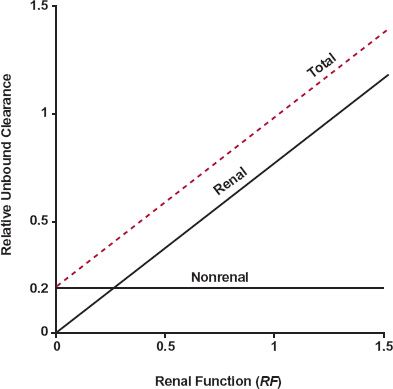
FIGURE 15-8. Renal, nonrenal, and total unbound clearances, expressed relative to the total unbound clearance of a typical patient, versus renal function (RF) for a drug with an fe(t) value of 0.8. Unbound nonrenal clearance remains constant and independent of renal function. In contrast, unbound renal clearance increases in direct proportion to renal function. The resulting (total) unbound clearance (colored dashed line) increases in parallel with renal clearance from a value of 0.2, represented by non-renal clearance, to a value of 1.0, when the renal function is that of the typical patient. In this illustration, renal function varies in a group of 60-year-old, 70-kg patients. Nonrenal clearance is, of course, expected to change with age and weight (Chapter 14, Age, Weight, and Gender). Note that RF values greater than 1.0 are shown to include those individuals with creatinine clearances greater than the average.
For a given age and weight, CLuNR(d) is expected to be constant, independent of renal function; whereas CLuR(d) is expected (Eq. 15-4) to change in direct proportion to renal function (RF). Thus, the unbound clearance in the patient, CLu(d), increases linearly with RF. The extent of the increase depends on fe(t), the contribution of the renal route to all routes of elimination in the typical patient. This relationship is shown in Fig. 15-8 for a drug with an fe(t) of 0.8.
On substituting Eqs. 15-1 to 15-5 into Eq. 15-6, the following useful relationship is derived for Rd, the unbound clearance ratio, CLu(d)/CLu(t).
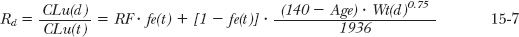
The first term, RF · fe(t), does not incorporate age and weight directly, because RF is estimated from creatinine clearance (next section) in the renally impaired patient relative to that of the typical patient. For a patient whose age and weight do not deviate far from that of a typical patient,
Rd = RF · fe(t) + 1 − fe(t) 15-8
Figure 15-9A illustrates how Rd, ratio of unbound clearances, depends on both the renal function remaining in a patient and the value of fe(t) in a typical 60-year-old, 70-kg patient. It is apparent that Rd changes the most with renal function when fe(t) = 1 and is unchanged when fe(t) = 0. The value of fe(t) is, by definition,
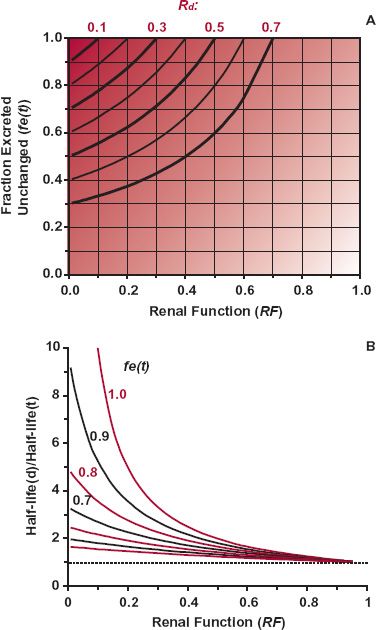
FIGURE 15-9. A. In patients with renal disease, the extent of decrease in Rd, the ratio of unbound clearances, depends on both the fraction excreted unchanged, fe(t) in typical patients and on the renal function (RF) remaining in an individual patient. The value of Rd is lowest (shown by deepest shading) when fe(t) approaches 1.0 and RF approaches zero. The lines show the combinations of fe(t) and RF that give the same Rd. B. The half-life of a drug, relative to the typical value, is expected to be the highest when RF approaches zero and fe(t) approaches 1.0. The lines indicate the ratio of half-lives for various values of fe(t) as a function of RF. Both panels are simulations based on Eq. 15-7 for a 70-kg, 60-year-old patient.
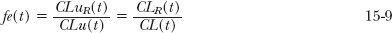
where CLuR(t) and CLu(t) are the unbound renal and total clearances, and CLR(t) and CL(t) are the corresponding values based on the total plasma concentration of the drug in the typical patient for whom the usual regimen is intended. Experimentally, fe(t) is the fraction of an i.v. dose recovered unchanged in urine in a typical patient.
Recall that 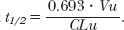 The half-life in a patient with renal function impairment, t1/2(d), compared with that in a typical patient with fully functioning kidneys, t1/2(t), is then
The half-life in a patient with renal function impairment, t1/2(d), compared with that in a typical patient with fully functioning kidneys, t1/2(t), is then

where Vu(d) and Vu(t) are the unbound volumes of distribution in the patient with renal disease and in the typical patient, respectively. Also, recall that Vu tends to vary in direct proportion to body weight. If renal function does not affect Vu, then

Figure 15-9B illustrates the dependence of half-life on both renal function and fraction excreted unchanged. Notice that half-life changes the most when renal function approaches zero and fe(t) approaches 1.0.
Before leaving this section, an additional point needs to be made with respect to data in Fig. 15-7 in which each point represents an observation from a different patient. The x-axis, creatinine clearance, is a function of age, weight, gender, and renal disease. One might expect that nonrenal clearance may also be related to age and weight. Higher values of nonrenal clearance occur in younger and larger patients, who have higher creatinine clearances, and smaller values are expected in older and smaller patients. This positive correlation of nonrenal clearance with creatinine clearance may distort the regression, so that the y-intercept is lower and the slope is greater than expected. Ideally, renal clearance should be correlated with creatinine clearance, and the nonrenal clearance should be examined separately for its dependence on age, weight, and perhaps renal disease.
DECREASE IN BINDING TO PLASMA PROTEINS
Renal disease is often associated with a decrease in binding to plasma proteins, especially those drugs that bind to albumin. The decreased binding is because of several factors, the most important of which is a decrease in albumin concentration. The accumulation of endogenous substances that may displace drug from the binding sites and the carbamylation of albumin (because of high levels of urea), that decrease its affinity for drugs, may also contribute. As a result of the decreased binding, clearance of metabolized drugs with a low hepatic extraction ratio tends to increase. Although the clearance of those drugs that are primarily excreted unchanged and bound to albumin also tends to increase, it is not as extensive as the decrease resulting from the decrease in renal function. Clearance of such drugs therefore decreases in renal disease, but it is only the unbound clearance that reflects unbound concentration, the correlate of drug response. This is the reason why the equations above (Eqs. 15-1 to 15-11) were derived using unbound drug.
ESTIMATION OF RENAL FUNCTION
Many methods have been developed over the years, since the physiology of the kidneys was originally worked out, to assess renal function. Two particularly useful methods that use serum creatinine are currently in common use.
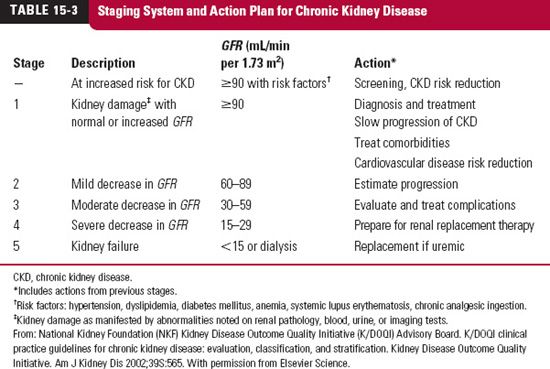
Estimation of Glomerular Filtration Rate. An estimate of glomerular filtration rate (GFR) is useful to assess the ability of the kidneys to handle drugs and other substances. Table 15-3 lists the GFR values used to define the stages of loss of renal function with age (see also Chapter 14, Age, Weight, and Gender) and concurrent diseases, such as diabetes and hypertension.
A method was recently developed using serum creatinine to determine GFR. GFR was directly assessed from the renal clearance of 125I-iothalamate. The procedure is known as the modification of diet in renal disease (MDRD) method. There are now two relationships used. The original procedure contains six variables; an adaptation, and now the more common one, contains four, as shown in Table 15-4. The calculated value is an estimate of the patient’s GFR and is normalized to 1.73 m2. Body weight is therefore not one of the variables. Although a valuable procedure for assessing renal function, it has not yet been extensively applied pharmacokinetically to adjust dosage in renal disease. Furthermore, for this purpose, GFR needs to be expressed in milliliters per minute, not milliliters per minute per 1.73 m2. Most of the literature to date on dosage adjustment uses creatinine clearance, a value which is typically about 15% to 20% greater than GFR.

Estimation of Creatinine Clearance. The usefulness of creatinine clearance lies in the observation that renal clearance of many drugs varies linearly with creatinine clearance (e.g., see Fig. 15-7), regardless of whether the drug is filtered only, secreted extensively, or reabsorbed in the renal tubule. Creatinine is virtually only eliminated from the body by renal filtration.
Under Chronic, Relatively Stable Conditions. In clinical practice, creatinine renal clearance is usually estimated from serum creatinine alone rather than from creatinine measurements in both plasma and urine. Apart from the extra analysis and inconvenience involved, incomplete urine collection is a major problem in the clinical setting often resulting in an underestimate of creatinine clearance. Serum creatinine alone is a reasonable indicator of renal function, because the daily production of creatinine is matched by its elimination under normal circumstances. Consequently, serum creatinine is related to creatinine clearance by
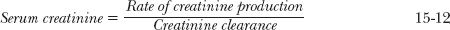
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


