OBJECTIVES
After studying this chapter, you should be able to:
Understand how nutrients are delivered to the body and the chemical processes needed to convert them to a form suitable for absorption.
List the major dietary carbohydrates and define the luminal and brush border processes that produce absorbable monosaccharides as well as the transport mechanisms that provide for the uptake of these hydrophilic molecules.
Understand the process of protein assimilation, and the ways in which it is comparable to, or converges from, that used for carbohydrates.
Define the stepwise processes of lipid digestion and absorption, the role of bile acids in solubilizing the products of lipolysis, and the consequences of fat malabsorption.
Identify the source and functions of short-chain fatty acids in the colon.
Delineate the mechanisms of uptake for vitamins and minerals.
Understand basic principles of energy metabolism and nutrition.
INTRODUCTION
The gastrointestinal system is the portal through which nutritive substances, vitamins, minerals, and fluids enter the body. Proteins, fats, and complex carbohydrates are broken down into absorbable units (digested), principally, although not exclusively, in the small intestine. The products of digestion and the vitamins, minerals, and water cross the mucosa and enter the lymph or the blood (absorption). The digestive and absorptive processes are the subject of this chapter.
Digestion of the major foodstuffs is an orderly process involving the action of a large number of digestive enzymes discussed in the previous chapter. Enzymes from the salivary glands attack carbohydrates (and fats in some species); enzymes from the stomach attack proteins and fats; and enzymes from the exocrine portion of the pancreas attack carbohydrates, proteins, lipids, DNA, and RNA. Other enzymes that complete the digestive process are found in the luminal membranes and the cytoplasm of the cells that line the small intestine. The action of the enzymes is aided by the hydrochloric acid secreted by the stomach and the bile secreted by the liver.
Most substances pass from the intestinal lumen into the enterocytes and then out of the enterocytes to the interstitial fluid. The processes responsible for movement across the luminal cell membrane are often quite different from those responsible for movement across the basal and lateral cell membranes to the interstitial fluid.
DIGESTION & ABSORPTION: CARBOHYDRATES
The principal dietary carbohydrates are polysaccharides, disaccharides, and monosaccharides. Starches (glucose polymers) and their derivatives are the only polysaccharides that are digested to any degree in the human gastrointestinal tract by human enzymes. Amylopectin, which typically constitutes around 75% of dietary starch, is a branched molecule, whereas amylose is a straight chain with only 1:4α linkages (Figure 26–1). The disaccharides lactose (milk sugar) and sucrose (table sugar) are also ingested, along with the monosaccharides fructose and glucose.
FIGURE 26–1
Left: Structure of amylose and amylopectin, which are polymers of glucose (indicated by circles). These molecules are partially digested by the enzyme amylase, yielding the products shown at the bottom of the figure. Right: Brush border hydrolases responsible for the sequential digestion of the products of luminal starch digestion (1, linear oligomers; 2, α-limit dextrins).
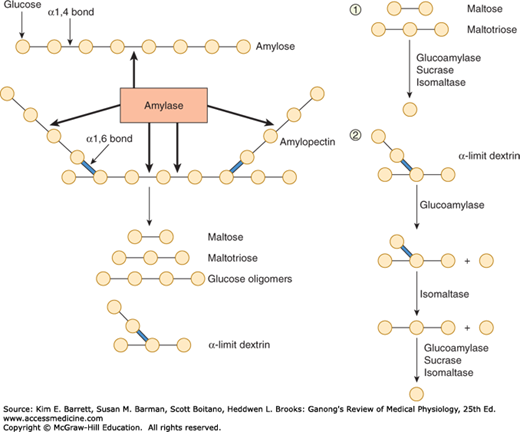
In the mouth, starch is attacked by salivary α-amylase. The optimal pH for this enzyme is 6.7. However, it remains partially active even once it moves into the stomach, despite the acidic gastric juice, because the active site is protected in the presence of substrate to some degree. In the small intestine, both the salivary and the pancreatic α-amylase also act on the ingested polysaccharides. Both the salivary and the pancreatic α-amylases hydrolyze internal 1:4α linkages but spare 1:6α linkages and terminal 1:4α linkages. Consequently, the end products of α-amylase digestion are oligosaccharides: the disaccharide maltose; the trisaccharide maltotriose; and α-limit dextrins, polymers of glucose containing an average of about eight glucose molecules with 1:6α linkages (Figure 26–1).
The oligosaccharidases responsible for the further digestion of the starch derivatives are located in the brush border of small intestinal epithelial cells (Figure 26–1). Some of these enzymes have more than one substrate. Isomaltase is mainly responsible for hydrolysis of 1:6α linkages. Along with maltase and sucrase, it also breaks down maltotriose and maltose. Sucrase and isomaltase are initially synthesized as a single glycoprotein chain that is inserted into the brush border membrane. It is then hydrolyzed by pancreatic proteases into sucrase and isomaltase subunits.
Sucrase hydrolyzes sucrose into a molecule of glucose and a molecule of fructose. In addition, lactase hydrolyzes lactose to glucose and galactose.
Deficiency of one or more of the brush border oligosaccharidases may cause diarrhea, bloating, and flatulence after ingestion of sugar (Clinical Box 26–1). The diarrhea is due to the increased number of osmotically active oligosaccharide molecules that remain in the intestinal lumen, causing the volume of the intestinal contents to increase. In the colon, bacteria break down some of the oligosaccharides, further increasing the number of osmotically active particles. The bloating and flatulence are due to the production of gas (CO2 and H2) from disaccharide residues in the lower small intestine and colon.
CLINICAL BOX 26–1 Lactose Intolerance
In most mammals and in many races of humans, intestinal lactase activity is high at birth, then declines to low levels during childhood and adulthood. The low lactase levels are associated with intolerance to milk (lactose intolerance). Most Europeans and their American descendants retain sufficient intestinal lactase activity in adulthood; the incidence of lactase deficiency in northern and western Europeans is only about 15%. However, the incidence in blacks, American Indians, Asians, and Mediterranean populations is 70–100%. When such individuals ingest dairy products, they are unable to digest lactose sufficiently, and so symptoms such as bloating, pain, gas, and diarrhea are produced by the unabsorbed osmoles that are subsequently digested by colonic bacteria.
THERAPEUTIC HIGHLIGHTSThe simplest treatment for lactose intolerance is to avoid dairy products in the diet, but this can sometimes be challenging (or undesirable for the individual who loves ice cream). Symptoms can be ameliorated by administration of commercial lactase preparations, but this is expensive. Yogurt is better tolerated than milk in intolerant individuals because it contains its own bacterial lactase.
Hexoses are rapidly absorbed across the wall of the small intestine (Table 26–1). Essentially all the hexoses are removed before the remains of a meal reach the terminal part of the ileum. The sugar molecules pass from the mucosal cells to the blood in the capillaries draining into the portal vein.
| Small Intestine | ||||
|---|---|---|---|---|
| Absorption of | Upperb | Mid | Lower | Colon |
| Sugars (glucose, galactose, etc) | ++ | +++ | ++ | 0 |
| Amino acids | ++ | ++ | ++ | 0 |
| Water-soluble and fat-soluble vitamins except vitamin B12 | +++ | ++ | 0 | 0 |
| Betaine, dimethylglycine, sarcosine | + | ++ | ++ | ? |
| Antibodies in newborns | + | ++ | +++ | ? |
| Pyrimidines (thymine and uracil) | + | + | ? | ? |
| Long-chain fatty acid absorption and conversion to triglyceride | +++ | ++ | + | 0 |
| Bile acids | + | + | +++ | + |
| Vitamin B12 | 0 | + | +++ | 0 |
| Na+ | +++ | ++ | +++ | +++ |
| K+ | + | + | + | Sec |
| Ca2+ | +++ | ++ | + | ? |
| Fe2+ | +++ | + | + | ? |
| Cl− | +++ | ++ | + | + |
| SO42− | ++ | + | 0 | ? |
The transport of glucose and galactose depends on Na+ in the intestinal lumen; a high concentration of Na+ on the mucosal surface of the cells facilitates sugar influx into the epithelial cells while a low concentration inhibits sugar influx into the epithelial cells. This is because these sugars and Na+ share the same cotransporter, or symport, the sodium-dependent glucose transporter (SGLT, Na+ glucose cotransporter) (Figure 26–2). The members of this family of transporters, SGLT-1 and SGLT-2, resemble the glucose transporters (GLUTs) responsible for facilitated diffusion (see Chapter 24) in that they cross the cell membrane 12 times and have their –COOH and –NH2 terminals on the cytoplasmic side of the membrane. However, there is no homology to the GLUT series of transporters. SGLT-1 is responsible for uptake of dietary glucose from the gut. The related transporter, SGLT-2, is responsible for glucose transport out of the renal tubules (see Chapter 37).
FIGURE 26–2
Brush border digestion and assimilation of the disaccharides sucrose (panel 1) and lactose (panel 2). Uptake of glucose and galactose is driven secondarily by the low intracellular sodium concentration established by the basolateral Na+, K+ ATPase (not shown). SGLT-1, sodium-glucose cotransporter-1.
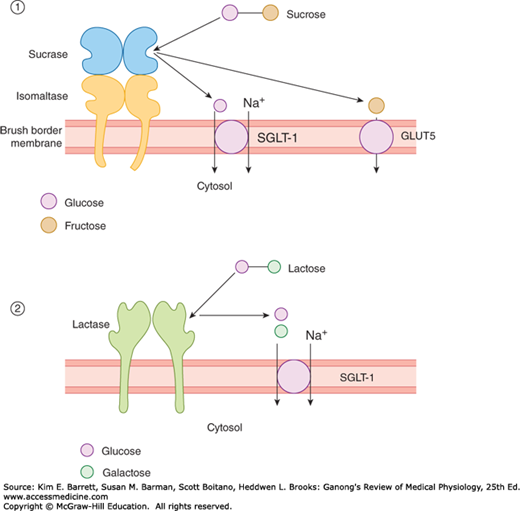
Because the intracellular Na+ concentration is low in intestinal cells (as it is in other cells), Na+ moves into the cell along its concentration gradient. Glucose moves with the Na+ and is released in the cell (Figure 26–2). The Na+ is transported into the lateral intercellular spaces, and the glucose is transported by GLUT2 into the interstitium and thence to the capillaries. Thus, glucose transport is an example of secondary active transport (see Chapter 2); the energy for glucose transport is provided indirectly, by the active transport of Na+ out of the cell. This maintains the concentration gradient across the luminal border of the cell, so that more Na+ and consequently more glucose enter. When the Na+/glucose cotransporter is congenitally defective, the resulting glucose/galactose malabsorption causes severe diarrhea that is often fatal if glucose and galactose are not promptly removed from the diet. Glucose and its polymers can also be used to retain Na+ in diarrheal disease, as was discussed in Chapter 25.
As indicated, SGLT-1 also transports galactose, but fructose utilizes a different mechanism. Its absorption is independent of Na+ or the transport of glucose and galactose; it is transported instead by facilitated diffusion from the intestinal lumen into the enterocytes by GLUT5 and out of the enterocytes into the interstitium by GLUT2. Some fructose is converted to glucose in the mucosal cells.
Insulin has little effect on intestinal transport of sugars. In this respect, intestinal absorption resembles glucose reabsorption in the proximal convoluted tubules of the kidneys (see Chapter 37); neither process requires phosphorylation, and both are essentially normal in diabetes but are depressed by the drug phlorizin. The maximal rate of glucose absorption from the intestine is about 120 g/h.
PROTEINS & NUCLEIC ACIDS
Protein digestion begins in the stomach, where pepsins cleave some of the peptide linkages. Like many of the other enzymes concerned with protein digestion, pepsins are secreted in the form of inactive precursors (proenzymes) and activated in the gastrointestinal tract. The pepsin precursors are called pepsinogens and are activated by gastric acid. Human gastric mucosa contains a number of related pepsinogens, which can be divided into two immunohistochemically distinct groups, pepsinogen I and pepsinogen II. Pepsinogen I is found only in acid-secreting regions, whereas pepsinogen II is also found in the pyloric region. Maximal acid secretion correlates with pepsinogen I levels.
Pepsins hydrolyze the bonds between aromatic amino acids such as phenylalanine or tyrosine and a second amino acid, so the products of peptic digestion are polypeptides of very diverse sizes. Because pepsins have a pH optimum of 1.6–3.2, their action is terminated when the gastric contents are mixed with the alkaline pancreatic juice in the duodenum and jejunum. The pH of the intestinal contents in the duodenal bulb is 3.0–4.0, but rapidly rises; in the rest of the duodenum it is about 6.5.
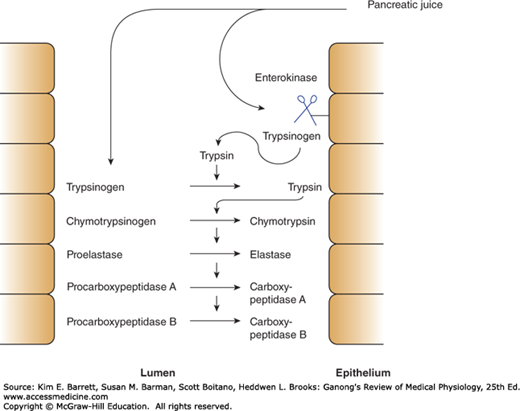
In the small intestine, the polypeptides formed by digestion in the stomach are further digested by the powerful proteolytic enzymes of the pancreas and intestinal mucosa. Trypsin, the chymotrypsins, and elastase act at interior peptide bonds in the peptide molecules and are called endopeptidases. The formation of the active endopeptidases from their inactive precursors occurs only when they have reached their site of action, secondary to the action of the brush border hydrolase, enterokinase (Figure 26–3). The powerful protein-splitting enzymes of the pancreatic juice are secreted as inactive proenzymes. Trypsinogen is converted to the active enzyme trypsin by enterokinase when the pancreatic juice enters the duodenum. Enterokinase contains 41% polysaccharide, and this high polysaccharide content apparently prevents it from being digested itself before it can exert its effect. Trypsin converts chymotrypsinogens into chymotrypsins and other proenzymes into active enzymes (Figure 26–3). Trypsin can also activate trypsinogen; therefore, once some trypsin is formed, there is an auto-catalytic chain reaction. Enterokinase deficiency occurs as a congenital abnormality and leads to protein malnutrition.
FIGURE 26–3
Mechanism to avoid activation of pancreatic proteases until they are in the duodenal lumen. Pancreatic juice contains proteolytic enzymes in their inactive, precursor forms. When the juice enters the duodenal lumen, trypsinogen contacts enterokinase expressed on the apical surface of enterocytes. Trypsinogen is thereby cleaved to trypsin, which in turn can activate additional trypsin molecules as well as the remaining proteolytic enzymes.
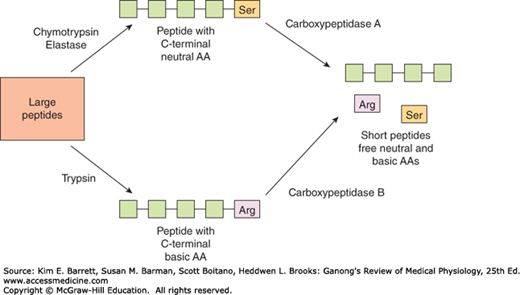
The carboxypeptidases of the pancreas are exopeptidases that hydrolyze the amino acids at the carboxyl ends of the polypeptides (Figure 26–4). Some free amino acids are liberated in the intestinal lumen, but others are liberated at the cell surface by the aminopeptidases, carboxypeptidases, endopeptidases, and dipeptidases in the brush border of the mucosal cells. Some dipeptides and tripeptides are actively transported into the intestinal cells and hydrolyzed by intracellular peptidases, with the amino acids entering the bloodstream. Thus, the final digestion to amino acids occurs in three locations: the intestinal lumen, the brush border, and the cytoplasm of the mucosal cells.
At least seven different transport systems transport amino acids into enterocytes. Five of these require Na+ and cotransport amino acids and Na+ in a fashion similar to the cotransport of Na+ and glucose (Figure 26–3). Two of these five also require Cl–. In two systems, transport is independent of Na+.
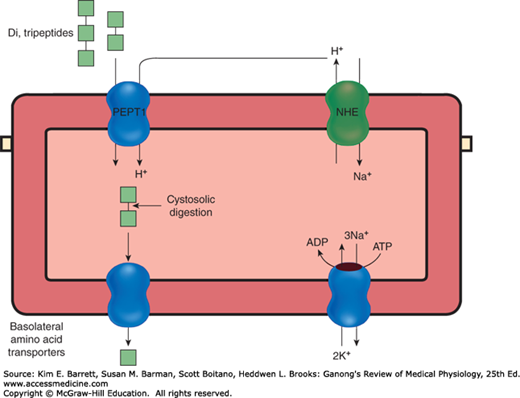
The dipeptides and tripeptides are transported into enterocytes by a system known as PepT1 (or peptide transporter 1) that requires H+ instead of Na+ (Figure 26–5). There is a very little absorption of larger peptides. In the enterocytes, amino acids released from the peptides by intracellular hydrolysis plus the amino acids absorbed from the intestinal lumen and brush border are transported out of the enterocytes along their basolateral borders by at least five transport systems. From there, they enter the hepatic portal blood.
FIGURE 26–5
Disposition of short peptides in intestinal epithelial cells. Peptides are absorbed together with a proton supplied by an apical sodium/hydrogen exchanger (NHE) by the peptide transporter 1 (PepT1). Absorbed peptides are digested by cytosolic proteases, and any amino acids that are surplus to the needs of the epithelial cell are transported into the bloodstream by a series of basolateral transport proteins.
Absorption of amino acids is rapid in the duodenum and jejunum. There is a little absorption in the ileum in health, because the majority of the free amino acids have already been assimilated at that point. Approximately 50% of the digested protein comes from ingested food, 25% from proteins in digestive juices, and 25% from desquamated mucosal cells. Only 2–5% of the protein in the small intestine escapes digestion and absorption. Some of this is eventually digested by bacterial action in the colon. Almost all of the protein in the stools is not of dietary origin but comes from bacteria and cellular debris. Evidence suggests that the peptidase activities of the brush border and the mucosal cell cytoplasm are increased by resection of part of the ileum and that they are independently altered in starvation. Thus, these enzymes appear to be subject to homeostatic regulation. In humans, a congenital defect in the mechanism that transports neutral amino acids in the intestine and renal tubules causes Hartnup disease. A congenital defect in the transport of basic amino acids causes cystinuria. However, most patients do not experience nutritional deficiencies of these amino acids because peptide transport compensates.
In infants, moderate amounts of undigested proteins are also absorbed. The protein antibodies in maternal colostrum are largely secretory immunoglobulins (IgAs), the production of which is increased in the breast in late pregnancy. They cross the mammary epithelium by transcytosis and enter the circulation of the infant from the intestine, providing passive immunity against infections. Absorption is by endocytosis and subsequent exocytosis.
Absorption of intact proteins declines sharply after weaning, but adults still absorb small quantities. Foreign proteins that enter the circulation provoke the formation of antibodies, and the antigen–antibody reaction occurring on subsequent entry of more of the same protein may cause allergic symptoms. Thus, absorption of proteins from the intestine may explain the occurrence of allergic symptoms after eating certain foods. The incidence of food allergy in children is said to be as high as 8%. Certain foods are more allergenic than others. Crustaceans, mollusks, and fish are common offenders, and allergic responses to legumes, cows’ milk, and egg white are also relatively frequent. However, in most individuals food allergies do not occur, and there is an evidence for a genetic component in susceptibility.
Absorption of protein antigens, particularly bacterial and viral proteins, takes place in large microfold cells or M cells, specialized intestinal epithelial cells that overlie aggregates of lymphoid tissue (Peyer patches). These cells pass the antigens to the lymphoid cells, and lymphocytes are activated. The activated lymphoblasts enter the circulation, but they later return to the intestinal mucosa and other epithelia, where they secrete IgA in response to subsequent exposures to the same antigen. This secretory immunity is an important defense mechanism (see Chapter 3).
Nucleic acids are split into nucleotides in the intestine by the pancreatic nucleases, and the nucleotides are split into the nucleosides and phosphoric acid by enzymes that appear to be located on the luminal surfaces of the mucosal cells. The nucleosides are then split into their constituent sugars and purine and pyrimidine bases. The bases are absorbed by active transport. Families of equilibrative (ie, passive) and concentrative (ie, secondary active) nucleoside transporters have recently been identified and are expressed on the apical membrane of enterocytes.
LIPIDS
A lingual lipase is secreted by Ebner glands on the dorsal surface of the tongue in some species, and the stomach also secretes a lipase (Table 26–1). They are of little quantitative significance for lipid digestion other than in the setting of pancreatic insufficiency, but they may generate free fatty acids that signal to most distal parts of the gastrointestinal tract (eg, causing the release of CCK; see Chapter 25).
Most fat digestion therefore begins in the duodenum, pancreatic lipase being one of the most important enzymes involved. This enzyme hydrolyzes the 1- and 3-bonds of the triglycerides (triacylglycerols) with relative ease but acts on the 2-bonds at a very low rate, so the principal products of its action are free fatty acids and 2-monoglycerides (2-monoacylglycerols). It acts on fats that have been emulsified (see below). Its activity is facilitated when an amphipathic helix that covers the active site like a lid is bent back. Colipase, a protein with a molecular weight of about 11,000, is also secreted in the pancreatic juice, and when this molecule binds to the –COOH-terminal domain of the pancreatic lipase, opening of the lid is facilitated. Colipase is secreted in an inactive proform (Table 26–1) and is activated in the intestinal lumen by trypsin. Colipase is also critical for the action of lipase because it allows lipase to remain associated with droplets of dietary lipid even in the presence of bile acids.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


