44 Diabetes mellitus
Diabetes mellitus is the most common of the endocrine disorders. It is a chronic condition, characterised by hyperglycaemia and due to impaired insulin secretion with or without insulin resistance. Diabetes mellitus may be classified according to aetiology, by far the most common types being type 1 and type 2 diabetes (Box 44.1). More than 2.6 million people in the UK have diabetes, and by the year 2025, this number is estimated to rise to 4 million.
Box 44.1 Aetiological classification of diabetes mellitus
Type 1 (β-cell destruction, usually leading to absolute insulin deficiency)
Type 2 diabetes is more common above the age of 40, with a peak age of onset in developed countries between 60 and 70 years, although it is being increasingly seen in younger people and even children. The prevalence of type 2 diabetes varies widely in different populations, being six times more common in those of South Asian origin compared with those of Northern European origin. It is caused by a relative insulin deficiency and insulin resistance. Symptoms are generally slower in onset and less marked than those of type 1. Type 2 diabetes may be an incidental finding, particularly when patients present with complications associated with the disease, for example, heart disease. Type 2 disease often progresses to the extent whereby extrinsic insulin is required to maintain blood glucose levels. The differences between type 1 and type 2 diabetes are highlighted in Table 44.1. It is sometimes difficult to distinguish clinically between type 1 and type 2 diabetes. The important thing to be aware of is that it is predominantly the degree of metabolic abnormality that is the key determinant of the form of treatment.
Table 44.1 Differences between type 1 and type 2 diabetes
| Type 1 diabetes | Type 2 diabetes |
|---|---|
| β-cell destruction | No β-cell destruction |
| Islet cell antibodies present | No islet cell antibodies present |
| Strong genetic link | Very strong genetic link |
| Age of onset usually below 30 | Age of onset usually above 40 |
| Faster onset of symptoms | Slower onset of symptoms |
| Insulin must be administered | Diet control and oral hypoglycaemic agents often sufficient control |
| Patients usually not overweight | Patients usually overweight |
| Extreme hyperglycaemia causes diabetic ketoacidosis | Extreme hyperglycaemia causes hyperosmolar hyperglycaemic state |
Pathophysiology
Type 2 diabetes is also associated with the metabolic syndrome (or syndrome X), although the real relevance of this ‘syndrome’ continues to be debated in the literature (Khan et al., 2005). The metabolic syndrome is a group of risk factors commonly found in those with type 2 diabetes, including insulin resistance, glucose intolerance (type 2 diabetes or IGT), hyperinsulinaemia, hypertension, dyslipidaemia, central obesity, atherosclerosis and increased levels of procoagulant factors, for example, plasminogen activator inhibitor-1 and fibrinogen.
Diagnosis
In June 2000, the UK formally adopted the World Health Organization criteria for diagnosing diabetes mellitus that was initially published in 1999. It has since been updated and the diagnostic criteria have been reiterated (World Health Organization, 2006).
Diabetic emergencies
Hypoglycaemia
Hypoglycaemia can occur both with insulin treatment and in those taking some oral agents, especially the longer-acting sulphonylureas, for example, chlorpropamide and glibenclamide. Definitions of hypoglycaemia vary, and in particular, there is no WHO definition. However, symptoms caused by the release of counter-regulatory hormones predominantly adrenaline (epinephrine), noradrenaline (norepinephrine) and glucagon tend to occur when the venous serum glucose drops below 3.0 mmol/L in healthy individuals. These symptoms described in Box 44.2 are a normal physiological response to hypoglycaemia and should alert the person to consume carbohydrates. Individuals may not respond appropriately to hypoglycaemia of this degree for several reasons, termed hypoglycaemia unawareness. First, the relevance of the symptoms has not been explained to them. This is an educational failing. It is imperative, therefore, that people with diabetes who are prescribed medication which is known to cause hypoglycaemia should be educated about the autonomic symptoms so that they may take action to avoid further decline of serum glucose. Second, the symptoms simply may not occur because of autonomic neuropathy. One of the commonest complications of diabetes is neuropathy, and when this includes the autonomic nervous system, there are no reliable symptoms to warn the individual that they are hypoglycaemic. A similar situation may occur as a consequence of drugs which suppress autonomic symptoms, such as β-blockers. Third, the patient may have recurrent hypoglycaemia. In those individuals who suffer frequent hypoglycaemic episodes, the autonomic symptoms may cease to occur. There is some evidence that the symptoms can be regained if, for a period of a few weeks, the serum glucose level can be maintained out of the hypoglycaemic range. Finally, the individual may be hypoglycaemia unaware because of alcohol intoxication.
Causes of hypoglycaemia
The most common causes of hypoglycaemia are either a decrease in carbohydrate consumption, excess carbohydrate utilisation from unexpected exercise or increase in circulating insulin (Table 44.2).
Table 44.2 Causes of hypoglycaemia
| Cause | Comment |
|---|---|
| Missed meals or delays in eating | Reduced carbohydrate intake, therefore reduction in glucose levels |
| Not eating the usual amount of carbohydrates | Reduced carbohydrate intake, therefore reduction in glucose levels |
| Increased doses of insulin | Increased uptake of glucose into cells and increased storage of glucose as glycogen |
| Increased doses of oral insulin secretagogues | Increased levels of insulin therefore increased uptake of glucose into cells and increased storage of glucose as glycogen |
| Introduction of other blood glucose-lowering agents to oral insulin secretagogues | Enhanced hypoglycaemic effects |
| Increase in exercise | Increased uptake of glucose into cells |
| Excessive alcohol consumption | Impaired gluconeogenesis |
| Liver disease | Impaired gluconeogenesis and glycogenolysis |
Long-term diabetic complications
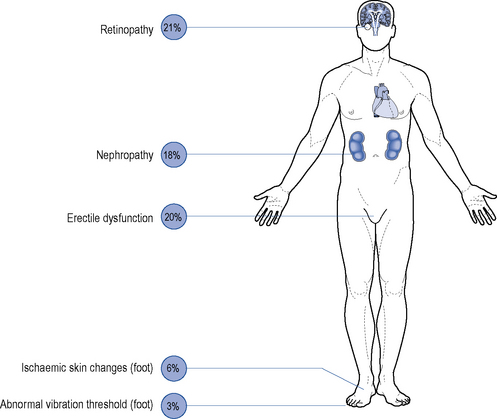
Although all long-term complications may occur in each type of diabetes, the spectrum of incidence is different. Many patients with type 2 diabetes have had their disease a long time before the diagnosis, by which time many have developed diabetic complications (Figs. 44.1 and 44.2). However, diabetic complications can be limited and sometimes prevented altogether if good management occurs from an early stage. Hyperglycaemia and hypertension are the two major modifiable risk factors that influence the development of diabetic complications.
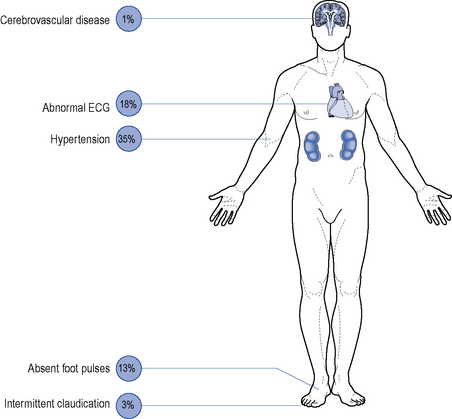
Macrovascular disease
Microvascular disease
Microvascular complications include retinopathy, nephropathy and neuropathy.
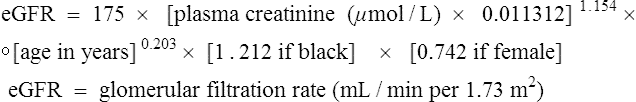
Peripheral neuropathy
Peripheral neuropathy is the progressive loss of peripheral nerve fibres resulting in nerve dysfunction. Diabetic neuropathies can lead to a wide variety of sensory, motor and autonomic symptoms. The most common is the symmetrical distal sensory type, which is particularly evident in the feet and may slowly progress to a complete loss of feeling. It is most prevalent in elderly patients with type 2 diabetes but may be found with any type of diabetes, at any age beyond childhood. Painful diabetic neuropathy is another manifestation of sensory neuropathy; it can be extremely disabling and may cause considerable morbidity. Guidance on the treatment of painful neuropathy is available (National Institute for Health and Clinical Excellence, 2010). Diabetic proximal motor neuropathy is rapid in onset and involves weakness and wasting, principally of the thigh muscles. Muscle pain is common and may require opiate analgesia. Distal motor neuropathy can lead to symptoms of impaired fine co-ordination of the hands and/or foot slapping.



