Chapter Fifty-One. Developmental anatomy
related cardiovascular and respiratory disorders
Introduction
This chapter considers common neonatal cardiovascular and respiratory problems. Specialist textbooks such as those by Taeusch et al (2005) and McCance & Huether (2007) and current research give fuller accounts of a wider range of pathophysiological developments and relevant clinical management. Neonates become ill very quickly and all neonates that cause concern should be referred to skilled paediatricians or a neonatal specialist so that any evolving problems are managed in a proactive manner thus minimising short- and long-term problems.
Aspects of cardiovascular development
The embryology of the anatomical structures of the heart is explained in Chapter 11 and the student should refer to the appropriate section. Of importance also is the conducting system of the heart. Prior to the septation of the atria and the ventricles, specialised conducting cells form the sinoatrial ventriculobulbar and bulbotruncal junctions. This specialised conduction tissue is thought to originate from the cardiac myocytes, possibly in response to specialised, although not as yet understood, signals (Bristow 2003). The looping of the primitive heart allows the atrioventricular ring to invaginate so that it lies at the base of the atrial septum; this allows some of the specialised conduction cells to establish continuity between the atrioventricular node and the bundle of His. Failure in the various segments of these specialised conducting cells to make contact with each other causes congenital heart block.
Cardiovascular problems
Cardiovascular abnormalities
Cardiovascular malformations make up the largest group (approx. 30%) of congenital anomalies. This averages about 8% per 1000 live births (Flanagan et al 1999). Neonates may be born with acyanotic or cyanotic cardiovascular anomalies which will require expert intervention. Although the specific cause of cardiac anomalies is unknown, the occurrence of many of them is linked with chromosomal and genetic aberrations and poorly understood syndromes. For example, atrioventricular and endocardial cushion defects are associated with trisomy 21—Down syndrome. The affected chromosomes/genes in some syndromes have been identified as in trisomy 21, genetic mutations encoding for extracellular matrix proteins, fibrillin-1 being responsible for Marfan syndrome and elastin being responsible for Williams syndrome. Genetic mutations on chromosome 22q11 may contribute to aortic arch anomalies, including Fallot’s tetralogy. As many of these anomalies are potentially life threatening, parents will seek an explanation for these problems and any risks for re-occurrence in future pregnancies.
In general, cardiovascular anomalies are grouped according to the direction of blood flow; thus defects that cause ‘ right-to-left shunts’ in the heart or great vessels almost always cause central cyanosis. In contrast, defects that cause ‘ left-to-right shunts’ in the heart or great vessels initially allow the neonate to be acyanotic (Table 51.1). Some cardiovascular anomalies cause obstruction, inhibiting either the right or the left ventricular outflow tract as seen in neonates with pulmonary and aortic valve hypoplasias. Those neonates that develop heart failure eventually present with considerable peripheral and central cyanosis.
| Right-to-left shunt | % | Left-to-right shunt | % | Obstructive disease | % |
|---|---|---|---|---|---|
| Transposition of the great vessels | 4 | Ventricular septal defect | 33 | Aortic stenosis | 6 |
| Tricuspid atresia | 1–2 | Patent ductus arteriosus | 10 | Pulmonary stenosis | 8 |
| Total anomalous pulmonary venous drainage | 1–2 | Atrial septal defect | 8 | Coarctation of the aorta | 6 |
Risk factors
Contributory factors thought to play a role in the development of some of the cardiovascular anomalies are:
• Maternal diabetes mellitus: ventricular septal defect (VSD), coarctation of the aorta and transposition of the great vessels.
• Chromosomal abnormalities such as Down syndrome (40%): atrioventricular defect (AVD), Fallot’s tetralogy, VSDs and patent ductus arteriosus (PDA).
• Genetic problems such as Williams syndrome.
• Family history, where siblings or a parent have a cardiac defect.
• Infectious agents such as those causing rubella or toxo-plasmosis infection in pregnancy.
• Nutritional/vitamin deficiencies such as folic acid deficiency (Chien 2000).
• Environmental factors and maternal drug abuse.
Presenting features in a neonate
Some of the following features may be present in neonates with cardiovascular anomalies although some of these features may also be suggestive of respiratory problems:
• Tachypnoea/dyspnoea.
• Grunting.
• Pulmonary plethora.
• Peripheral/central cyanosis.
• Tachycardia.
• Heart murmurs.
• Poor feeding ability.
• Poor weight gain and growth retardation.
• Hepatomegaly.
• Cachexia.
A paediatrician must examine all neonates presenting with any of the above symptoms; however, as some cardiovascular symptoms can be vague the actual anomaly may not be diagnosed for several days or weeks and usually following extensive investigations in a specialist centre.
Investigations
Persistent tachypnoea may be the first indication of a cardiovascular or pulmonary anomaly. Generally, cardiovascular anomalies that cause excessive pulmonary arterial blood flow and pulmonary venous hypertension enlarge the pulmonary vessels causing pulmonary oedema, decreasing the neonate’s lung compliance. The cumulative effects of all these events manifest in significant respiratory difficulties and cyanosis. In contrast, cardiovascular anomalies resulting in a decrease in pulmonary blood flow tend to contribute to intense cyanosis that elicits tachypnoea without significant respiratory distress. A persistent respiratory rate of 60 breaths/min or greater accompanied by increased respiratory depth commonly precedes clinical deterioration.
Chest radiograph
A chest X-ray will establish the size and position of the heart in relation to the lungs and mediastinum as some cardiovascular defects and anomalies alter the normal size, shape and position of the heart. Any deviations from the normal appearance of the mediastinum, the heart, greater blood vessels and lungs contribute to the diagnosis.
Electrocardiography
Electrocardiography is extremely valuable when neonates present with cardiac arrhythmia of any origin. ECG recordings often reflect the degree of neonatal adaptation to extrauterine life. Specialist practitioners should interpret the recorded evidence, distinguishing between the gradual transition in right ventricular dominance noticeable in the first 72 h and electrophysiological anomalies that may help towards the overall diagnostic assessment.
Echocardiography
Exploration of the cardiovascular system by two-dimensional echocardiography involves ultrasound scanning (Fig. 51.1) and contributes to the analysis of cardiovascular and intracardiac structures (Knight & Washington 2006). Neonates are good candidates for echocardiographic imaging because of their thin thoracic walls. Images of the heart chambers, and valves and dimensions of the large vessels can be easily identified. Although many abnormalities can be identified in the fetus from 16 weeks of gestation, new images are required after birth to confirm diagnosis.
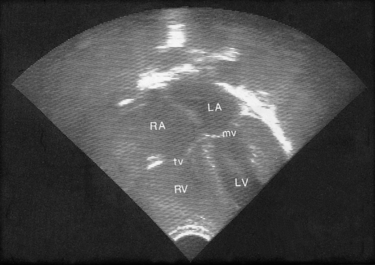 |
| Figure 51.1 A four chamber echo of the normal heart. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle; tv, tricuspid valve; mv, mitral valve. (From Kelnar C, Harvey D, Simpson C 1995, with permission.) |
Colour images help to visualise the presence and direction of blood flow in neonates with persistent patent ductus arteriosus, septal defects, systemic venous anomalies and arteriovenous malformations. Pulsed and continuous-wave Doppler techniques can estimate pressure gradients across stenotic valves, septal defects and abnormal vessel structures such as the dimensions of patent ductus arteriosus and coarctation of the aorta. Palliative and corrective surgery is sometimes initiated solely on the basis of echocardiographic findings, without more invasive investigations such as cardiac catheterisation, but such decisions are dependent on the nature and severity of the cardiovascular anomaly.
Magnetic resonance imaging
MRI detects intrathoracic abnormalities of the aortic arch, the peripheral pulmonary arteries and the systematic collateral vessels, which are not adequately evaluated by echocardiography.
Cardiac catheterisation and angiocardiography
These are invasive procedures that are required in neonates with complex cardiovascular defects when explicit details are needed about the size of the defects and related haemodynamics. A fine catheter is inserted into the femoral vein, inferior vena cava, and the right side of the heart, the pulmonary artery and its branches. In general, such procedures have diagnostic purposes although occasionally cardiac catheterisation can be used to provide palliative treatment for neonates born with transposition of the great arteries or tricuspid atresia, whose life is dependent on interatrial blood flow. Then the catheterisation is carried out under general anaesthesia and the intracardiac catheter is used to carry out a balloon atrial septostomy which enhances systemic and pulmonary blood flow. If a septal defect is present, it may be possible to pass the catheter into the left side of the heart. Abnormal tracts can be identified and blood pressure and oxygen saturation within the heart and great vessels can be measured (Feinstein & Moore 2003).
Arterial blood gases
Measurement of arterial blood gases from a baby breathing air followed by 100% oxygen can provide evidence for a differential diagnosis of a right-to-left shunt in cyanotic heart disease. Po 2 seldom exceeds 25 kPa in a baby with cyanotic heart disease breathing 100% oxygen, whereas a baby with cyanosis caused by lung disorders will usually have a higher level of arterial oxygen. However, this test may provoke closure of the ductus arteriosus with disastrous consequences in neonates dependent on ductal blood flow.
Some common disorders: acyanotic lesions
Patent ductus arteriosus
The incidence of isolated persistent patency of the ductus arteriosus is about 10–12% of all congenital heart disease (Friedman & Silverman 2001), although Hoffman (2003)suggests a lower percentage at 7.1%. It is twice as common in females as in males. In a term neonate the ductus arteriosus usually closes within the first few hours of birth in response to increased blood oxygen saturation. This functional closure is then followed by an anatomical occlusion several days later. Premature neonates may take up to 3 months to achieve such spontaneous closures which are attributed to the development of specialised contractile smooth muscle tissue and intimal thickening and fibrosis.
Where the patency of the ductus arteriosus persists, the fall in the neonate’s pulmonary resistance reverses the direction of blood flow through the ductus from the aorta (left) shunting the oxygenated blood back into the pulmonary circulation (right). In some neonates the ductus arteriosus remains open when cardiac anomalies such as pulmonary stenosis are present (Fig. 51.2). In such instances the additional pulmonary blood flow is advantageous until the anomaly is surgically corrected. In the absence of the more complex cardiac anomalies, failure of spontaneous closure of the ductus arteriosus in premature neonates can contribute to persistence of respiratory distress syndrome (RDS). In term neonates it may lead to congestive cardiac failure, pulmonary hypertension and subacute bacterial endocarditis.
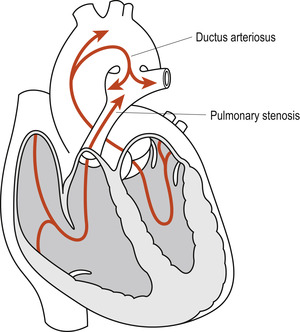 |
| Figure 51.2 Persistent ductus arteriosus in the presence of pulmonary stenosis. (From Fitzgerald M J T, Fitzgerald M 1994, with permission.) |
Symptoms of persistent ductus arteriosus develop between the 3rd and 7th days following birth. The neonate develops tachypnoea, dyspnoea and lethargy. Systolic and diastolic murmurs are almost always present. Therapeutic intervention includes restriction of fluids, adequate oxygenation and, where necessary, treatment for congestive cardiac failure (CCF) with digitalis, furosemide ( frusemide) and potassium supplementation.
Indometacin may be used to induce ductus closure in premature neonates although term neonates are not as responsive to this drug, possibly due to its quicker clearance through the kidneys (Clyman 2005). As indometacin binds to plasma albumin, its presence may displace bilirubin and contribute to jaundice. Where appropriate expertise is available, transcatheter device occlusion may be the method of choice for ductal closure; however, more invasive methods of closure by means of a thoracotomy or thoracoscopy (Hoffman 2003) which facilitate ligation may be necessary if the earlier medical management fails.
Ventricular septal defects
Ventricular septal defects (VSDs) are common and account for 20% of congenital heart malformations (Friedman & Silverman 2001). A VSD can occur as an isolated intracardiac abnormality or it can be an adjunct to other cardiovascular lesions as is the case in about 50% of neonates born with congenital cardiovascular abnormalities. The defect usually occurs in the membranous septum but can present anywhere within the muscular part of the interventricular septum (Fig. 51.3).
 |
| Figure 51.3 Ventricular septal defect. (From Fitzgerald M J T, Fitzgerald M 1994, with permission.) |
The size and complexity of such defects varies from minute openings to almost complete absence of the interventricular septum. Knowledge of the precise size and position of the defect(s) is critical to successful therapeutic management. The incidence of spontaneous closure of the muscular defect is high, averaging 70%, although surgical repair is required where larger defects fail to close or membranous defects persist. Infants with small defects may be asymptomatic and eventually experience spontaneous closure, resulting in normal cardiac anatomy.
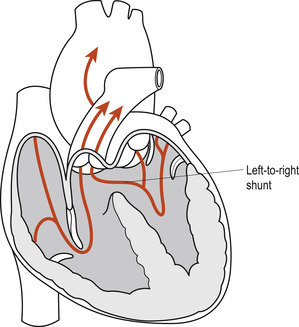
Larger defects are likely to lead to CCF, feeding problems and delays in growth. For this reason, VSDs are classified as acyanotic heart lesions with increased pulmonary blood flow caused by the increasing left ventricular pressure occurring after birth. The defect generates a left-to-right interventricular flow of blood. The presence of multiple VSDs or a large VSD allows the left-to-right shunt to equalise the pressure in both ventricles due to the shunting of oxygenated blood from the left ventricle into the right ventricle. This shunting will eventually lead to right ventricular volume overload, pulmonary plethora and CCF. Furthermore, the persistent increase in pulmonary blood volume increases pulmonary venous return to the left side of the heart. This eventually results in left ventricular volume overload, exacerbation of the left-to-right shunt and left ventricular hypertrophy.
For a time, the enlarged left ventricle pumps more efficiently, but eventually the heart fails and pulmonary hypertension develops. If untreated, the persistent problems contribute to the development of Eisenmenger’s syndrome (McCance & Huether 2007). Pre-emptive therapeutic interventions are important if long-term problems are to be averted or minimised. Early diagnosis and proactive management of CCF as above should be considered until the defect is closed surgically. Where necessary, surgery is delayed until the infant is at least 12–18 months old but all defects must ideally be closed, surgically if necessary, by the time the child enters school.
Atrial septal defects
Atrial septal defects are relatively common congenital cardiac malformations occurring in 1:1000 of live births (Wernovsky & Gruber 2005). Interferences with the sequential atrial septal development may cause a variety of atrial septal defects (Srivastava & Olson 2000) some of which may be simple, involving only the foramen ovale, while others may be complex. Severe atrioventricular defects may extend to the ventricular septum, the mitral and tricuspid valves.
The most common of these defects are:
• Septum primum with endocardial cushion defects. This defect is generally associated with abnormalities of the mitral and tricuspid valves and atrioventricular canals (Fig. 51.4A). Although this defect may occur in isolation in otherwise normal neonates, 30% of cases occur in neonates with Down syndrome in whom the involvement of the endocardial cushions may also contribute to the defective formation of the upper portion of the intraventricular septum.
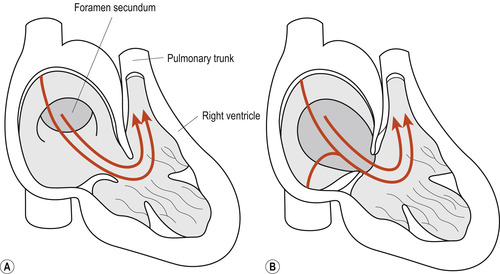 |
| Figure 51.4 (A) Septum primum lesion. (B) Ostium secundum lesion. (From Fitzgerald M J T, Fitzgerald M 1994, with permission.) |
• Ostium secundum. This defect is found in the central portion of the atrial septum. Because of its close anatomical association with the fossa ovalis, it is often referred to as the fossa ovalis defect (Fig. 51.4B).
• Sinus venosus defect. This defect is characteristically found in the superior portion of the atrial septum and generally extends into the superior vena cava.
Simple defects cause few problems and, if spontaneous closure does not occur, surgical repairs are carried out by about 4–5 years of age. As these defects cause a left-to-right intracardiac shunt, no cyanosis occurs. However, for neonates born with atrioventricular defects (AVD), the prognosis is less favourable as they may experience conduction problems. Complex atrial septal defects are usually diagnosed in the first few days of life as these neonates develop tachypnoea, mild cyanosis, abnormal heart sounds and systolic murmurs, poor feeding and later fail to gain weight (Rudolph et al 2003).
Some common disorders: cyanotic lesions
Transposition of the great arteries
Complete transposition of the great arteries (TGA) is the most common cardiovascular cause of peripheral and central cyanosis in neonates; the aorta arises from the right ventricle and the pulmonary artery from the left ventricle. This complete switch of the great vessels (Fig. 51.5) leads to the formation of two separate closed circulatory systems, whereby the blood from the pulmonary circulation cannot enter the systemic circulation and vice versa. Survival of these neonates depends on the patency of the right-to-left shunts in the ductus arteriosus and/or the foramen ovale. The presence of a concurrent VSD also permits mixing of the oxygenated and deoxygenated blood at the interventricular level. Boys appear to be affected twice as often as girls (Flanagan et al 1999).
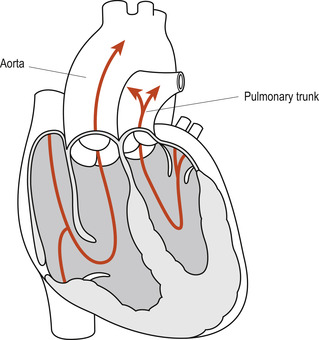 |
| Figure 51.5 Transposition of great arteries. (From Fitzgerald M J T, Fitzgerald M 1994, with permission.) |
A neonate with complete TGA and no accompanying ventricular defect will become increasingly cyanotic as the ductus arteriosus begins to close. This cyanosis is not relieved by administration of 100% oxygen and urgent life-saving intervention is needed. This is likely to consist of prostaglandin infusion to maintain the PDA open until palliative or corrective surgery can be carried out.
The palliative intervention may consist of a balloon septostomy (e.g. Rashkind’s), generally carried out as an emergency to enlarge the foramen ovale and increase the interatrial mixing of blood. When possible, corrective surgery will be carried out some weeks/months later (Fig. 51.6). This consists of switching the pulmonary artery and the aorta to the correct ventricles and closing any remaining cardiovascular defects. Care is taken to ensure that the coronary arteries can sustain normal myocardial perfusion. Most surgical corrections are successful with a survival rate of up to 90% (McCance & Huether 2007).
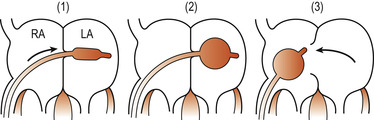 |
| Figure 51.6 Rashkind’s atrial septostomy. (1) A catheter is passed into the right atrium (RA) and pushed through the foramen ovale into the left atrium (LA). (2) A balloon at the tip of the catheter is inflated. (3) The catheter is withdrawn sharply so that the inflated balloon tears the atrial septum, allowing oxygenated blood to reach the systemic circulation. (Reproduced with permission from Wallis & Harvey 1979.) |
Total anomalous pulmonary venous drainage
Total anomalous pulmonary venous connection occurs in 1:150 000 and is characterised by the absence of any direct connection of the pulmonary veins to the left atrium. Instead, the pulmonary veins connect either to the right atrium, various systemic veins or the liver. Depending on these unusual connections, oxygenated blood is returned into the right atrium, via the superior vena cava, the coronary sinus or the ductus venosus. About 30% of neonates born with total anomalous pulmonary venous drainage have other cardiac anomalies that must be considered in the overall management. Almost all surviving neonates present with a right-to-left shunt of blood at atrial or ventricular level, allowing mixed arterial and venous blood to circulate to the lungs and body. Central cyanosis, dyspnoea, tachypnoea and CCF occur and urgent, life-saving surgical correction is necessary. Mortality can be between 5% and 25% (Knight & Washington 2006).
Tetralogy of Fallot
The tetralogy of Fallot is the most common form of cyanotic congenital lesion in infants beyond the first year of life. Neonates can sometimes be asymptomatic. The four anatomic defects are (Fig. 51.7):
1. A high, large VSD.
3. Pulmonary stenosis: a funnel-shaped opening at the entrance to the pulmonary artery. The pulmonary artery and valve may rarely be completely obliterated.
4. Right ventricular hypertrophy, developing because of the obstruction to blood flow.
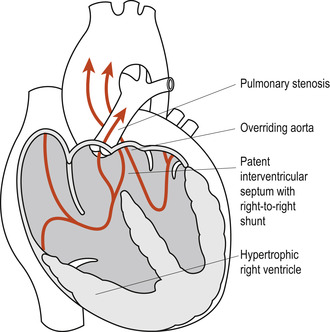 |
Figure 51.7
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|