, Sam Salek2 and Stuart Walker3
(1)
Centre of Regulatory Excellence, Duke-NUS Graduate Medical School, Singapore, Singapore
(2)
Department of Pharmacy, University of Hertfordshire, Hatfield, UK
(3)
Centre for Innovation in Regulatory Sciences, London, UK
Introduction
Currently, there is a need to understand why different regulatory agencies come to different outcomes despite having the same data submitted for their assessment. This has led to an increasing pressure on agencies to improve transparency and accountability and establish appropriate document governance for their decision-making processes. A universal framework (CMR 2008) would be of value and should be applicable to both pharmaceutical companies and regulatory agencies resulting in a standardized framework for benefit–risk assessment to support transparency in decision-making.
A survey conducted within pharmaceutical companies and regulatory agencies showed that the main hurdle to establishing a universal framework was the lack of an accepted, validated, and international model. It is therefore vital to establish a universal framework with the participation of major regulatory agencies to ensure the possible uptake of the same framework by other regulators across the world. One of the challenges is to harmonize the different requirements of such a framework for the assessment of benefits and risks of medicines which could be applied across different jurisdictions and scenarios.
At a time of constrained resources, shared and joint reviews are a possible way forward, and this led to the formation of the Consortium, consisting of four similar-sized agencies (Swissmedic, Therapeutic Goods Administration (TGA), Singapore’s Health Sciences Authority (HSA), and Health Canada). The four agencies had a plan to initiate work sharing whereby a harmonized benefit–risk assessment template would be required. In order to achieve this, it was important to review the existing frameworks and select one for further development.
Objectives
This study had the following objectives, namely, to develop:
1.
A universal framework for benefit–risk assessment of medicines to achieve a systematic approach to benefit–risk decision-making
2.
A benefit–risk template to document benefit–risk decision-making using the benefit–risk framework principles
3.
A user manual for regulatory assessors to guide the use of the Benefit–Risk Template
Methods
In order to develop and propose a universal framework that facilitated decision-making, the expectations and requirements of such a framework were obtained through a review of published literature and reports from relevant workshops. Opinions were then collated and organized to provide a list of requirements for a universal framework for the benefit–risk assessment of medicines.
Existing frameworks for the assessment of benefits and risks of medicines were reviewed. The selected frameworks were assessed against the list of criteria which included logical soundness, comprehensiveness, acceptability of results, practicality, specificity and sensitivity, presentation (visualization), and scope. Finally, the selected framework was evaluated by comparing the components with those of existing frameworks to determine if it included the essential elements for a universal framework.
Benefit–risk decisions need to be communicated in an effective and systematic manner, allowing appropriate understanding of the information by the stakeholders. A template should be an aid for documenting the processes leading to the construction of a benefit–risk balance and the eventual basis that would support the decision. A search was conducted for guidances used by regulatory agencies in order to identify those elements considered essential to the assessment of benefits and risks of a medicine. The EMA guidance document of 2008 was utilized in developing an appropriate BR template. These elements were then transformed into a template that allowed documentation and editing. This initial developmental template was then reviewed against the universal framework so that it could support the principles outlined in the overarching universal framework.
The initial template was assessed by the Consortium who evaluated its use in a feasibility study, and the template was amended and finalized based on the feedback from the Consortium. Comments from the reviewers of the template highlighted the need for a user manual. It was found that the usefulness of the template would be dependent on an understanding of the terms and requirements of the input fields and compliance in completing the template. The Consortium identified areas in the template that would require clarification or additional explanation. These provided the critical elements in producing the user manual to guide users in completing the template. The initial user manual was further revised by the Consortium resulting in the final version.
Results
The results are presented in three parts, namely:
Part I: Development of the universal framework
Part II: Development of the Benefit–Risk Template
Part III: Development of the User Manual
Part I: Development of the Universal Framework
Requirements of a Universal Framework
The EMA Benefit–Risk Methodology Project (EMA 2009) was aimed at the development and testing of tools and processes for balancing multiple benefits and risks, which could be used as an aid to informed, science-based regulatory decisions about medicinal products. This project consisted of five consecutive work packages. The second work package (EMA 2010) examined the applicability of three qualitative frameworks, namely, the Pharmaceutical Research and Manufacturers of America (PhRMA) Benefit–Risk Action Team framework (BRAT framework), the seven-step framework developed by the Centre for Innovation in Regulatory Science (CIRS), and the benefit–risk framework developed by the US FDA, and the 18 quantitative approaches for assessing the benefit–risk balance.
It was found that clinical judgment remained a critical role in regulatory decision-making and models could assist but not replace the complex process of constructing a benefit–risk balance and incorporating uncertainties into the final decision. In the EMA’s evaluation of quantitative approaches, it was concluded that any quantitative method or approach would require a qualitative framework within which the model could be effectively developed. Combinations of approaches could prove useful in situations that required a review of the contributions by the magnitude of favorable effects, seriousness of unfavorable effects, uncertainties, transitions in health states and the time spent in each state, and trade-offs between effects. Therefore, an overarching benefit–risk assessment framework with the capacity to incorporate various quantitative methods would be ideal.
The International Society for Pharmacoeconomics and Outcomes Research (ISPOR) Risk–Benefit Management Working Group conducted a study (Guo et al. 2010) to review and compare published quantitative benefit–risk assessment methodologies employed by regulatory agencies and/or the pharmaceutical industry in the hope that comparisons may help disclose unique characteristics of the techniques that may be more applicable to a specific drug evaluation scenario or a specific therapeutic indication. It was found that each quantitative method had its unique advantages and disadvantages based on data requirements and statistical properties. Numerous methodologies have been proposed, but there were a limited number of empirical applications of these techniques, and there was no consensus among regulators for defining a clear gold standard. When evaluating any new healthcare technology, Guo et al. 2010) recommended the use of multiple benefit–risk assessment approaches across different therapeutic indications and treatment populations to construct the benefit–risk profile. This was similar to the EMA opinion regarding the need to vary the tools available for effective benefit–risk assessment, which should be governed under an overarching framework.
In the report of methods for benefit and harm assessment in systematic reviews by the Agency for Healthcare Research and Quality (Boyd et al. 2012), some principles for a review protocol development were highlighted. Firstly, the key potential benefits and harms should be identified. Then, the approaches used in the reporting of the benefit and harm outcomes should be indicated, including the assumptions undertaken for the approaches described, e.g., number needed to treat (NNT) and number needed to harm (NNH). This would help to understand the appropriateness and rationale for the approaches selected. Preferences (including patients’ preferences) should also be considered in the assessment and sensitivity analyses conducted to determine the impact of varying preferences. In delivering the overall benefit–risk assessment, a qualitative or quantitative approach should be clearly stated.
Mussen et al. (2009) conducted a literature review of tools for the assessment of medicines and argued that the development of a new model ought to achieve the following objectives:
1.
Framework should match current practices of regulatory agencies for benefit–risk assessment, in order that the framework can be used in the scope of those practices.
2.
Framework should be able to take into account the data in a marketing authorization application and the scientific data otherwise available to regulatory agencies.
3.
Framework should not require additional analyses or reanalyses of source clinical data or additional clinical meta-analyses.
4.
Framework should be used for initial registration and post-approval reassessment of existing medicines.
5.
Framework should be applicable to all kinds of medicines, including vaccines and nonprescription medicines.
6.
Framework should be considered a tool for regulatory agencies and pharmaceutical companies for assessing benefit–risk balance of medicines but not substitute decision-making.
7.
Framework should be validated.
A study was conducted to explore the current status and the need for a universal benefit–risk framework for medicines in regulatory agencies and pharmaceutical companies (Chap. 3). It was found that for the utility and scope of a universal benefit–risk assessment framework, most agencies and companies believed that a benefit–risk framework should be applied throughout the life cycle of the medicine with the emphasis on applicability to product registration, health technology assessment agencies, and across the life cycle of a product (Table 4.1). The general consensus was that a benefit–risk framework should be utilized by both agencies and companies. Both agencies and companies also believed that a universal framework would enhance the quality of communication and enable the assessment of benefit–risk management plans. The advantages of a universal framework were as follows: it would provide documentation for a structured discussion, act as a tool for communication among peers within the organization, and enable communication between the organization and stakeholders. There was a general agreement that these advantages would include appropriate documentation and enhancement of communication together with transparency and accountability of decisions.
Table 4.1
Requirements of a universal benefit–risk framework
Utility and scope of a universal framework |
Need for a universal benefit–risk assessment framework |
Importance of a universal benefit–risk framework developed for registration purposes |
Importance of a universal benefit–risk framework applied throughout the life cycle of a medicine |
Applicability of a universal benefit–risk framework to health technology assessment agencies |
Utility of a universal benefit–risk assessment framework |
Purposes of a universal framework |
Application of a universal benefit–risk framework to benefit–risk management plans |
Transparency and consistency of decision-making |
Communication of decision |
Chapter 3 also identified the criteria from both agencies and companies for reviewing a benefit–risk framework (Table 4.2). These would be used to assess the suitability of frameworks in consideration for further development into a universal framework. The findings from EMA and Guo et al. (2010) for an overarching framework allowing various assessment tools can be subsumed under the criterion “comprehensiveness.”
Table 4.2
Criteria influencing the quality of a universal benefit–risk framework
1. Logical soundness | Provides an approach that is sound and allows decisions that are coherent and aids rational thinking |
2. Comprehensiveness | Provides an approach that handles all forms of data (including qualitative and quantitative and subjective and objective information) and allows for multiple criteria |
3. Acceptability of results | Provides an approach that checks for inconsistencies in data and judgment and a realistic approach to the evaluation of benefits and risks |
4. Practicality | Provides an approach with minimum burden on resources and ease of use |
5. Specificity and sensitivity | Provides a statistical perspective underpinning the reliability of the decision |
6. Presentation (visualization) | Provides outcomes in an easily understandable format such as charts and plots |
7. Scope | Provides a consistent approach throughout drug development and post-approval monitoring |
Identification of a Suitable Framework
There were five frameworks identified that are currently used for the assessment of the benefits and risks of medicines (Table 4.3). Of these, two were used by regulatory agencies and another two by pharmaceutical companies. The seven-step framework by CIRS had been reviewed by both the major stakeholders, namely, regulatory agencies and pharmaceutical companies. None were currently used as a universal framework.
Table 4.3
Frameworks currently used for the assessment of benefits and risks of medicines
Source | CIRS | EMA | US FDA | PhRMA | Novo Nordisk |
|---|---|---|---|---|---|
Name of framework | Seven-step framework | Eight-step PrOACT-URL | Five-step benefit–risk framework | Six-step BRAT framework | Eight-step BRAIN framework |
Basis of framework | MCDA | MCDA | MCDA | MCDA | |
Reviewed by | Regulatory agencies and pharmaceutical companies | EU regulatory agencies | US regulatory agency | Pharmaceutical companies | Pharmaceutical companies |
Multi-criteria decision analysis (MCDA) was the platform on which other frameworks were based, and it was also confirmed as a useful relevant methodology (Chap. 3). MCDA is a process described in the Multi-Criteria Analysis (MCA) manual (Dodgson et al. 2009) which aims to explore the individual contributing aspects of the decision-making process before collating the outcomes to form the basis of the decision. There are three key phases of the MCDA process. The problem is first identified and structured, and secondly the decision maker’s preferences are taken into account. Lastly, action plans are developed. The steps in executing these three key phases can be found in Table 4.4.
Table 4.4
Steps in multi-criteria decision analysis (MCDA)
Steps | Actions | |
|---|---|---|
1. | Establish the decision context | Establish aims of the MCDA; identify decision makers and other key players |
Design the socio-technical system for conducting the MCDA | ||
Consider the context of the appraisal | ||
2. | Identify the options to be appraised | |
3. | Identify objectives and criteria | Identify criteria for assessing the consequences of each option |
Organize the criteria by clustering them under high-level and lower-level objectives in a hierarchy | ||
4. | Scoring – assess the expected performance of each option against the criteria, then assess the value associated with the consequences of each option for each criterion | Describe the consequences of the options |
Score the options on the criteria | ||
Check the consistency of the scores on each criterion | ||
5. | Weighting – assign weights for each of the criteria to reflect their relative importance to the decision | |
6. | Combine the weights and scores for each option to derive an overall value | Calculate the overall weighted scores at each level in the hierarchy |
Calculate the overall weighted scores | ||
7. | Examine the results | |
8. | Sensitivity analysis | Conduct a sensitivity analysis: do other preferences or weights affect the overall ordering of the options? |
Look at the advantage and disadvantage of the selected options and compare pairs of options | ||
Create possible new options that might be better than those originally considered | ||
Repeat the above steps until a “requisite” model is obtained |
An important feature of the MCDA model is the ability to carry out sensitivity analyses on the results by varying any of the weights and scores to assess the impact on the overall benefit–risk balance. The MCDA model generates two assessments of the data, with the first being the overall value (cumulative outcomes after scoring and weighting) and the second a sensitivity analysis (through adjusting the scores and weights). The criteria to be taken into account in determining the outcome for the assessment were grouped as “benefits” and “risks.” The criteria for risks included not only the incidence of adverse events and drug-related reactions, but also unobserved and potential risks based on knowledge of factors including related products and the mechanism of action.
Each criterion would then be assigned a score and given a weight according to its relative importance to the benefit–risk decision. Weighted scores were then calculated at each level in the hierarchy which enabled an overall weighted score to be calculated for each of the options. The process of “scoring” would be based predominantly on measurable data such as the clinical trial endpoints and incidence of adverse events, measured as percentages. The process of “weighting” the criteria was where experience and judgment were built into the methodology. The assignment of weight to a criterion was normally based on a combination of factors on which a value judgment would be made.
MCDA is believed to have the following advantages as it:
Takes explicit account of multiple and conflicting criteria
Helps to structure the problem
Helps decision makers learn about the problem and their own and others’ values and judgment and, through structuring and presenting the information, identify a preferred course of action
Serves to complement and challenge intuition, but does not seek to replace intuitive judgment or experience
Leads to better considered, justified, and explainable decisions and provides an audit trail
Demonstrates that decisions are conceptually simple and transparent
In addition, in support of a universal benefit–risk framework, the MCDA model is not limited by type of data and is used for approval or post-marketing and with all types of medicines. It makes use of available data without the need to conduct further analyses and does not aim to replace decision-making but provides clarity with respect to the basis of the decision made. Scoring, weighting, and sensitivity analyses fulfill the requirements for a universal framework that could check for inconsistencies in the data (acceptability of results) as well as specificity and sensitivity.
MCDA, in providing a structured flow of information leading to a decision, is a tool for communicating a transparent and consistent decision. It also appears not be limited in its scope and can be applied to benefit–risk management plans and be used by health technology assessment agencies.
The factors influencing the quality of a universal benefit–risk framework were reviewed against the MCDA approach and the steps in executing this model. The structure of MCDA, in presenting and organizing information, provides logical soundness, and since it uses available data, it would be a comprehensive and practical framework not limited by the scope of application in approval and post-marketing scenarios. However, MCDA does not provide any form of visualization that could enhance the ease of understanding the outcomes. It could help enhance the consistency, objectivity, and transparency of the decision-making process for benefit–risk assessments by providing a structured and systematic approach and appropriate documentation for tracking the process and providing greater accountability. It also facilitates the reviewing of past decisions and experiences to ensure the consistency of regulatory decisions on marketing authorization applications. Through this, a better understanding could be achieved of the contexts as to why different agencies could reach different conclusions on the basis of the same data as well as imparting objectivity to the regulatory process.
It thus appeared that frameworks using the MCDA approach could be considered appropriate for further development into a universal framework. The CIRS seven-step framework was chosen as the model for further development into a universal framework due to its independent development and its exposure to both regulatory agencies and pharmaceutical companies.
Development of the Framework
The CIRS seven-step framework, based on the three key phases of MCDA, was reviewed to identify areas of improvement. The processes of this seven-step framework are described in Fig. 4.1. Step 1, namely, “decision context,” is the identification and structuring of the problem, while steps 2–5 are the development of decision maker’s preferences, i.e., criteria for benefits and risks. Step 7 is “expert judgment” and correlated to the final key phase of MCDA in providing an action plan leading to a decision. It should be noted that step 6 “visual presentation” was added to fulfill the requirements as identified earlier for a universal framework.
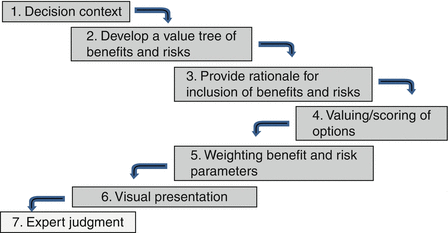
Fig. 4.1
The initial seven-step framework for the assessment of benefits and risks of medicines
Although the CIRS seven-step framework had been reviewed by both major stakeholders, it had not been applied in the real-world situation. Noting that groups of the four other frameworks were currently used individually by the respective developers, harmonization of the essential elements was conducted to impart a character of universal utility to the CIRS framework. This would help incorporate the existing work processes of the various stakeholders around the world and make the potential uptake of the universal framework more appropriate.
The US FDA used a framework (Table 4.5) that would accurately and concisely describe benefit and risk considerations to help assessors apply a structured approach in regulatory decision-making (CIRS 2011). An important consideration is the context of the decision, an understanding of the condition treated, and the unmet medical need. A more systematic and open discussion with informed patients could provide valuable insights in a given disease and the potential gaps or limitations in available therapies. There are now ongoing projects to develop and implement a plan to integrate a benefit–risk framework in the drug review process during PDUFA V (FDA 2012a).
Table 4.5
US FDA’s five-step approach to assessment of benefits and risks
Consideration | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
Analysis of condition | Summary of evidence | Conclusions (implications for decision) |
Unmet medical need | Summary of evidence | Conclusions (implications for decision) |
Clinical benefit | Summary of evidence | Conclusions (implications for decision) |
Risk | Summary of evidence | Conclusions (implications for decision) |
Risk management | Summary of evidence | Conclusions (implications for decision)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|