Fig. 22.1 The actions of drugs used in the treatment of depression on CNS serotonergic and adrenergic functioning.
The primary action of many drugs in current clinical use is to enhance serotonin (5-HT, 5-hydroxytryptamine) and noradrenaline (NA) availability. The majority of released serotonin and noradrenaline is rapidly removed from the synapse by reuptake into the neuron (yellow circles). Antidepressants vary in their abilities to inhibit the reuptake of serotonin or noradrenaline, thus enhancing the synaptic concentrations of these transmitters. Stimulation of presynaptic α2-adrenoceptors reduces monoamine release; mirtazapine, by blocking these presynaptic autoreceptors, increases noradrenaline and serotonin release and transmission. Other drugs act by significantly blocking postsynaptic receptors which are upregulated in depression. βAR, β-adrenoceptor; MAO, monoamine oxidase; MAOI, monoamine oxidase inhibitor; NA, noradrenaline; NRI, (selective) noradrenaline reuptake inhibitor; SNRI, serotonin and noradrenaline reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant.
In contrast, most noradrenergic neurons arise in the locus coeruleus and the lateral tegmental areas of the brainstem. The locus coeruleus and the raphe region have many reciprocal neural projections, and therefore the pathways are interdependent. For example, noradrenergic neurotransmission stimulates serotonergic neurons by activating somatodendritic α1-adrenoceptors, but also inhibits serotonin synthesis and release through presynaptic α2-adrenoceptors.
Pathways mediated by glutamate, γ-aminobutyric acid (GABA) and substance P also modulate monoaminergic neurotransmission.
Monoamine neurotransmitters and depression
Serotonergic pathways in the CNS are believed to be mainly involved in mood, while noradrenergic pathways are involved in stress systems, drive and energy state. These monoaminergic circuits in the brain are closely integrated. Simplistically, it has been hypothesised that the following biological changes in the monoamine system are important in depression:



There are increased 5-HT2 receptor numbers in the frontal cortex of depressed suicide victims, whereas other studies have indicated that serotonin and noradrenaline concentrations in the brain are reduced in depression. Overall, evidence for the ‘monoamine’ theory as a molecular basis for depression is limited, but the response to drugs that increase monoamine neurotransmission supports the concept.
Regulation of brain-derived neurotrophic factor in depression
Most antidepressant drugs increase the CNS monoamine concentrations rapidly, but the clinical benefit of antidepressant therapy is delayed. This suggests that more gradual adaptive changes occur as a result of increased monoaminergic neurotransmission. These pharmacologically induced changes are incompletely understood, but they may help to normalise the fundamental dysfunction in intracellular signalling pathways and transduction mechanisms that have been described in depression.
There is evidence for the central role of brain-derived neurotrophic factor (BDNF) in depression. Regulation of BDNF by monoamines is shown in Figure 22.2. BDNF expression is reduced when monoamine neurotransmission is impaired, but also in conditions of stress with elevated serum cortisol. Decreased expression of BDNF has adverse effects on neuronal plasticity can reduce neuronal networks, and may be a major factor in loss of neuronal circuitry and hippocampal atrophy. There is some evidence that successful antidepressant treatment is associated with increased BDNF expression and a restoration of hippocampal function and neuroendocrine regulation.
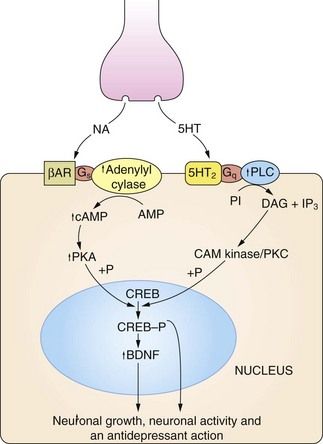
Fig 22.2 The regulation of neuronal growth and plasticity by monoamines and brain-derived neurotrophic factor (BDNF).
Adequate levels of monoamines, cAMP response element-binding protein (CREB-P) and BDNF are considered necessary for neuronal growth and plasticity. An increase in cAMP can result from noradrenaline (NA) acting on β-adrenoceptor (βAR) subtypes, and an increase in diacylglycerol (DAG) signalling can result from serotonin (5-HT) acting on 5-HT2 type receptors. There is also evidence that cAMP can be increased by serotonin acting on 5-HT4 and 5-HT7 receptors, and DAG by noradrenaline acting on α1-adrenoceptors (not shown). Critical points in this cascade may be dysfunctional in depressed individuals, including reduced synthesis of the monamine transmitters, genetic polymorphism affecting the function or expression of monoamine receptors, anomalies in coupling of Gs to adenylyl cyclase, reduced protein kinase A (PKA) activity and reduced phosphorylation of CREB. These may result in reduced BDNF activity, leading to neuronal atrophy and cell death in the hippocampus and cortex. CAM kinase, calmodulin-dependent protein kinase; IP3, inositol triphosphate; PI, phosphatidylinositol; PKC, protein kinase C; PLC, phospholipase C.
Antidepressant drug action
Most of the antidepressant drugs currently used clinically target the mechanisms involved in the control of monoamine neurotransmitter turnover or monoamine receptor function. There seems to be little difference in efficacy between drugs that act predominantly on serotonergic or on noradrenergic pathways, although they differ in their side-effect profiles. The ways that major antidepressants work to modify monoamine turnover and function are shown in Figure 22.1.
Long-term treatment with antidepressants promotes both the structural and functional integrity of the neural circuits that regulate mood. The mechanisms by which they achieve this are complex.
 Enhanced CNS monoamine levels. The initial action of most drugs used in the treatment of depression is to enhance neurotransmission by CNS monoamines, particularly serotonin but also noradrenaline and dopamine. Increased noradrenergic activity further enhances serotonergic neurotransmission by stimulating somatodendritic α1-adrenoceptors on serotonergic neurons. However, although antidepressants rapidly increase synaptic monoamine levels, clinical improvement is delayed. In part, this may be due to slow reduction in the number of upregulated somatodendritic and presynaptic 5-HT1 inhibitory autoreceptors (see above), which is necessary before activity increases in serotonergic pathways.
Enhanced CNS monoamine levels. The initial action of most drugs used in the treatment of depression is to enhance neurotransmission by CNS monoamines, particularly serotonin but also noradrenaline and dopamine. Increased noradrenergic activity further enhances serotonergic neurotransmission by stimulating somatodendritic α1-adrenoceptors on serotonergic neurons. However, although antidepressants rapidly increase synaptic monoamine levels, clinical improvement is delayed. In part, this may be due to slow reduction in the number of upregulated somatodendritic and presynaptic 5-HT1 inhibitory autoreceptors (see above), which is necessary before activity increases in serotonergic pathways. Effects on postsynaptic monoamine receptor expression and intracellular signal transduction. During treatment with antidepressants there is a gradual increase in responsiveness to serotonin in the prefrontal cortex. There is considerable evidence that antidepressants reverse the changes in intracellular signalling that are found in depression (Fig. 22.2). They enhance the response to monoamine receptor stimulation, which increases expression of BDNF and its receptor. As a result there is enhanced differentiation of progenitor cells into neurons and increased neuronal survival. Of note, electroconvulsive therapy can also increase the expression and activity of BDNF.
Effects on postsynaptic monoamine receptor expression and intracellular signal transduction. During treatment with antidepressants there is a gradual increase in responsiveness to serotonin in the prefrontal cortex. There is considerable evidence that antidepressants reverse the changes in intracellular signalling that are found in depression (Fig. 22.2). They enhance the response to monoamine receptor stimulation, which increases expression of BDNF and its receptor. As a result there is enhanced differentiation of progenitor cells into neurons and increased neuronal survival. Of note, electroconvulsive therapy can also increase the expression and activity of BDNF. Regulation of CRH production. During long-term treatment with antidepressants there is normalisation of overexpressed CRH secretion. This may be related to upregulation of CNS glucocorticoid receptors, with feedback inhibition of CRH.
Regulation of CRH production. During long-term treatment with antidepressants there is normalisation of overexpressed CRH secretion. This may be related to upregulation of CNS glucocorticoid receptors, with feedback inhibition of CRH.
Antidepressant drugs
Tricyclic antidepressant drugs
Mechanism of action: Tricyclic antidepressants (TCAs) inhibit the reuptake of monoamine neurotransmitters into the presynaptic neuron by competitive inhibition of monoamine transporter (MAT) proteins, particularly the noradrenaline transporter NET and the serotonin transporter SERT (Fig. 22.1). Some drugs show little monoamine selectivity, while other compounds are more selective for one monoamine (Table 22.1). However, the degree of monoamine selectivity has not been shown to influence efficacy. The subsequent effects on the CNS are described above.
Table 22.1
Comparative properties of some commonly used antidepressant drugs
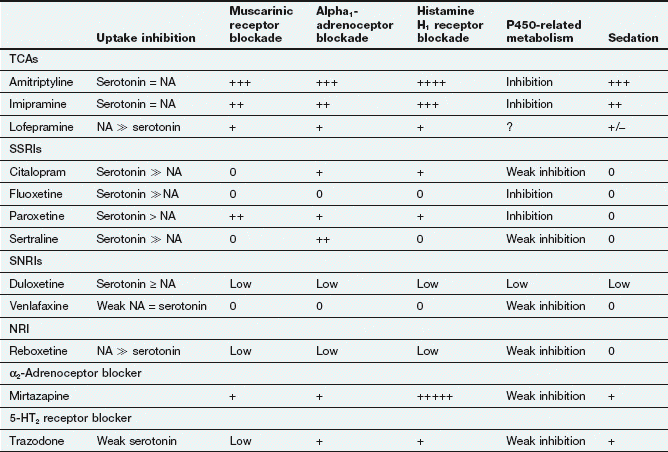
Drugs are listed under their conventional groupings but many have mixed or uncertain mechanisms of action. Differential blockade of muscarinic receptors, α1-adrenoceptors and histamine H1 receptors contributes to the side-effect profiles of antidepressant drugs. Other antidepressant drugs, including monoamine oxidase inhibitors (MAOIs), are listed in the Compendium at the end of the chapter. NA, noradrenaline; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin and noradrenaline reuptake inhibitor; NRI, noradrenaline reuptake inhibitor; TCA, tricyclic antidepressant.
The table is constructed from data in Richelson E (2002) The clinical relevance of antidepressant interaction with neurotransmitter transporters and receptors. Psychopharmacol Bull 36(4), 133–150 and other sources for approximate comparison only.
Many of the unwanted effects of these drugs are a consequence of blockade of other postsynaptic receptors (e.g. muscarinic and histamine H1 receptors and α1-adrenoceptors) (Table 22.1), which do not influence their antidepressant action.
Pharmacokinetics: All TCAs are well absorbed from the gut and highly protein bound in plasma. They undergo extensive first-pass metabolism in the liver, and active metabolites are formed which are partially responsible for the variable effective half-lives of these drugs (8–90 h; see Compendium at the end of this chapter). The combination of high first-pass metabolism and high clearance but a long elimination half-life is explained by high apparent volumes of distribution (10–50 L⋅kg−1 body weight). There is considerable inter-individual variability in the first-pass metabolism of most TCAs, leading to up to 40-fold differences in the plasma concentrations of the parent drug. There is no clear dose relationship for the therapeutic effects, although unwanted effects are dose-related. Dose titration is usually necessary to optimise the therapeutic response; this should be gradual over 1–2 weeks to minimise unwanted effects.
 Sedation as a result of histamine H1 receptor and α1-adrenoceptor blockade (Ch. 39). Some compounds are highly sedative, for example amitriptyline, and others less so. Sedation can be useful to help restore sleep patterns in depression (using a larger dose of a sedative drug at night) but can be troublesome or even dangerous during the day.
Sedation as a result of histamine H1 receptor and α1-adrenoceptor blockade (Ch. 39). Some compounds are highly sedative, for example amitriptyline, and others less so. Sedation can be useful to help restore sleep patterns in depression (using a larger dose of a sedative drug at night) but can be troublesome or even dangerous during the day. Antimuscarinic effects (see Ch. 4): dry mouth is a frequent occurrence, and less commonly constipation, urinary retention, impotence and visual disturbance. Tolerance to these effects can occur and gradual increases in dose may reduce their incidence.
Antimuscarinic effects (see Ch. 4): dry mouth is a frequent occurrence, and less commonly constipation, urinary retention, impotence and visual disturbance. Tolerance to these effects can occur and gradual increases in dose may reduce their incidence.
 Postural hypotension produced by peripheral α1-adrenoceptor blockade (Ch. 4) can be particularly troublesome in the elderly, although tolerance can occur.
Postural hypotension produced by peripheral α1-adrenoceptor blockade (Ch. 4) can be particularly troublesome in the elderly, although tolerance can occur. Epileptogenic effects: TCAs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history.
Epileptogenic effects: TCAs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history. Cardiotoxicity in overdose: most tricyclic drugs depress myocardial contractility. They can produce tachycardia and severe arrhythmias when taken in overdose, due to both antimuscarinic effects and excessive noradrenergic stimulation. Lofepramine is less cardiotoxic than other drugs in this class.
Cardiotoxicity in overdose: most tricyclic drugs depress myocardial contractility. They can produce tachycardia and severe arrhythmias when taken in overdose, due to both antimuscarinic effects and excessive noradrenergic stimulation. Lofepramine is less cardiotoxic than other drugs in this class.
 Hyponatraemia from inappropriate antidiuretic hormone (ADH; vasopressin) secretion, leading to drowsiness, confusion and convulsions.
Hyponatraemia from inappropriate antidiuretic hormone (ADH; vasopressin) secretion, leading to drowsiness, confusion and convulsions. Sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm.
Sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm. Sudden withdrawal syndrome: during long-term treatment doses should be gradually reduced over four weeks to avoid agitation, headache, malaise, sweating and gastrointestinal upset, which can accompany sudden withdrawal. These may result from excessive cholinergic activity following prolonged muscarinic receptor blockade.
Sudden withdrawal syndrome: during long-term treatment doses should be gradually reduced over four weeks to avoid agitation, headache, malaise, sweating and gastrointestinal upset, which can accompany sudden withdrawal. These may result from excessive cholinergic activity following prolonged muscarinic receptor blockade.Drug interactions: Several important drug interactions are recognised. TCAs potentiate the central depressant activity of many drugs, including alcohol. A dangerous interaction can result from giving a monoamine oxidase (MAO) inhibitor (MAOI) (see below) and a TCA together due to prolonged action of the increased serotonin released from the neuron. The interaction can lead to hyperpyrexia, convulsions and coma, and can occur up to two weeks after stopping an MAOI due to the long duration of MAO inhibition.
The risk of serious arrhythmias is increased when TCAs are taken with drugs that prolong the Q–T interval on the electrocardiogram (Ch. 8). Such drugs include the class III antiarrhythmic sotalol, and all class I antiarrhythmics.
Selective serotonin reuptake inhibitors and related antidepressants
Mechanism of action: Unlike the TCAs, the selective serotonin reuptake inhibitors (SSRIs) reduce the neuronal reuptake of serotonin by its presynaptic transporter protein (SERT), but have little or no effect on noradrenaline reuptake (Table 22.1). They have a more favourable profile of unwanted effects than TCAs because of their low affinity for muscarinic and histamine receptors and α1-adrenoceptors. Paroxetine is unusual among SSRIs in having affinity for muscarinic M3 receptors, found in the brain, salivary glands and smooth muscle.
The proposed mechanism of action of SSRIs is as follows.
 The increase in synaptic serotonin concentration as a result of reduced neuronal uptake leads to downregulation of the 5-HT1 somatodendritic and axon terminal presynaptic inhibitory autoreceptors on serotonergic neurons.
The increase in synaptic serotonin concentration as a result of reduced neuronal uptake leads to downregulation of the 5-HT1 somatodendritic and axon terminal presynaptic inhibitory autoreceptors on serotonergic neurons.
Subsequent changes in intracellular function are described above.
Pharmacokinetics: SSRIs are well absorbed from the gut and metabolised in the liver. Paroxetine has a long half-life (10–20 h), which is greatest in poor metabolisers of CYP2D6 substrates (30–50 h). Citalopram, fluoxetine and sertraline have very long half-lives (23–75 h). The active metabolite of fluoxetine has a half-life of 6 days, and the resulting very long duration of action can be a disadvantage if an MAOI is used subsequently (see below).
Unwanted effects: In contrast to the TCAs, SSRIs have few antimuscarinic effects (apart from paroxetine), cause little sedation or weight gain and are not cardiotoxic in overdose. However, they may cause:




 hyponatraemia, due to inappropriate secretion of ADH and leading to drowsiness, confusion and convulsions, is more frequent than with TCAs,
hyponatraemia, due to inappropriate secretion of ADH and leading to drowsiness, confusion and convulsions, is more frequent than with TCAs,
 sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm. This affects up to three-quarters of people taking SSRIs,
sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm. This affects up to three-quarters of people taking SSRIs,
 SSRIs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history,
SSRIs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history, sudden withdrawal syndrome after long-term use, which may be most troublesome with paroxetine; it presents with gastrointestinal symptoms, headache, anxiety, dizziness, paraesthesia, electric shock sensations in the head, neck and spine, sleep disturbance and sweating and usually begins 24–72 h after stopping treatment. The dose should be gradually reduced over at least 4 weeks,
sudden withdrawal syndrome after long-term use, which may be most troublesome with paroxetine; it presents with gastrointestinal symptoms, headache, anxiety, dizziness, paraesthesia, electric shock sensations in the head, neck and spine, sleep disturbance and sweating and usually begins 24–72 h after stopping treatment. The dose should be gradually reduced over at least 4 weeks,
Drug interactions: The most serious interaction is with MAOIs (see TCAs above). An interval of five weeks is recommended after stopping fluoxetine, or two weeks after paroxetine or sertraline, before an MAOI (including selegiline, Ch. 24) is taken. Fluoxetine and other SSRIs inhibit hepatic CYP2D6 (Table 2.7), and this can increase the plasma concentration of drugs metabolised by this enzyme.
Serotonin and noradrenaline reuptake inhibitors
Mechanism of action and uses: Venlafaxine and duloxetine are classified as serotonin and noradrenaline reuptake inhibitors (SNRIs) although at lower doses they have a greater effect on serotonin reuptake (Table 22.1). Like the TCAs, they inhibit neuronal reuptake of both serotonin and noradrenaline, but share with SSRIs a low affinity for muscarinic and histamine receptors and α1-adrenoceptors. Their unwanted effect profiles are therefore closer to those of the SSRIs than those of the TCAs. There is some evidence that clinical improvement with venlafaxine may begin earlier than with other antidepressant drugs.
Duloxetine is also used as an adjunctive treatment for smoking cessation (Ch. 54), and in urinary stress incontinence (Ch. 15).
Pharmacokinetics: Venlafaxine and duloxetine are well absorbed from the gut and undergo extensive first-pass metabolism in the liver. The main active metabolite of venlaxafine has a long half-life (11 h) and the half-life of duloxetine is 9–19 h.





 QT segment prolongation on the ECG with venlafaxine, which predisposes to ventricular arrhythmias (see Ch. 8); it should be avoided in people at high risk of arrhythmias.
QT segment prolongation on the ECG with venlafaxine, which predisposes to ventricular arrhythmias (see Ch. 8); it should be avoided in people at high risk of arrhythmias.Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


