Dense Deposit Disease
A. Brad Farris, III, MD
Key Facts
Terminology
C3 nephropathy with very electron-dense deposits within GBM and mesangium
Clinical Issues
Rare (1-3 cases/million)
Primarily children 5-15 years old, also adults
Proteinuria/hematuria, nephritic or nephrotic syndrome
C3NeF present in > 80%
No effective treatment
˜ 50% develop ESRD in 10-15 years
Recurs in renal allografts
Ocular drusen
Acquired partial lipodystrophy (APL) (˜ 5%)
Microscopic Pathology
Varied glomerular pathology
Mesangial proliferation, acute exudative GN, MPGN, crescentic GN
GBMs are thickened, eosinophilic, and refractile and stain strongly with PAS
IF: “Garland” and granular mesangial pattern of C3 in 100%
IgM (35%), IgG (25%), IgA (15%), or C1q (10%)
EM: Highly osmiophilic dense deposits within GBM, mesangium, Bowman capsule, and TBM
Deposits in Bruch membrane and spleen
Top Differential Diagnoses
AGN; MPGN, types I/III; MIDD
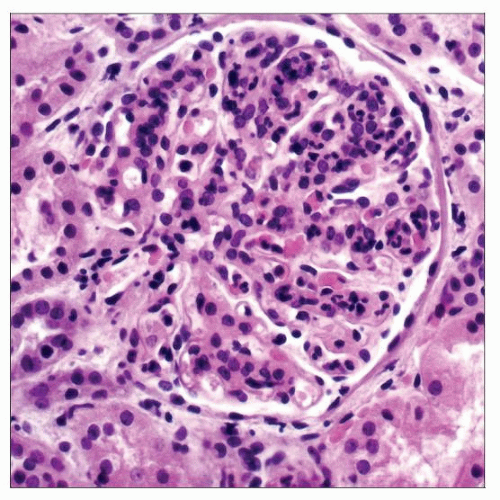 Dense deposit disease (DDD) often presents with an MPGN pattern, shown here in a biopsy from a 13-year-old boy with gross hematuria and proteinuria 3 days after a meningococcal septicemia. Serum C3 was undetectable. |
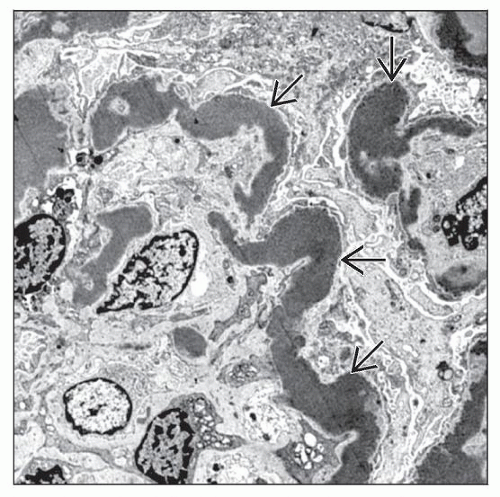 The diagnosis of DDD is made by electron microscopy, which reveals extremely dark, osmiophilic deposits along the GBM  . Deposits are found also in the mesangium, Bowman capsule, and TBM. . Deposits are found also in the mesangium, Bowman capsule, and TBM. |
TERMINOLOGY
Abbreviations
Dense deposit disease (DDD)
Synonyms
Membranoproliferative glomerulonephritis (MPGN), type II
Definitions
C3-related glomerulopathy manifested by broad, linear, extremely electron-dense deposits with C3 within GBM, mesangium, Bowman capsule, and TBM
Once classified as variant of MPGN, but MPGN pattern present in < 50%
Hyperdense deposits by EM are pathognomonic; therefore, DDD is preferred name
Initially reported by Galle and Berger in 1962
ETIOLOGY/PATHOGENESIS
Chronic Activation of Alternative Complement Pathway
Autoantibodies to complement components
C3 nephritic factor (C3NeF): Autoantibody to C3 convertase of alternative pathway (C3bBb) prevents inactivation, resulting in continuous activation of complement alternative pathway
C3NeF present in > 80% of patients (˜ 100% children, ˜ 40% adults)
C3NeF may also be present in healthy individuals
Specificity of antibody affects functional consequences
Autoantibodies to CFH, factor B, or C3
Mutations in complement component genes
Factor H mutations lead to low plasma levels or affect its C3bBb decay function
Tyrosine 402 to histidine common
C3 mutation resistant to factor H in fluid phase
Deposition of C3 in Lamina Densa of GBM
Local activation of complement pathway
Recruitment of leukocytes
Inflammatory damage of glomerular components
Loss of heparan sulfate (principal negative charge barrier)
Precipitating Factors
Infection, various (pneumonia, upper respiratory)
Group A streptococcal or meningococcal infections
Post chemotherapy for breast cancer
˜ 20% of adults with DDD have monoclonal gammopathy including myeloma
˜ 70% of patients have monoclonal gammopathy of undetermined significance
Some patients have multiple susceptibility factors
Animal Models
CFH mutation in Norwegian Yorkshire pigs
Mouse strain with CFH knockout
Prevented by combined factor B or factor I knockout
Proves necessity of alternative pathway convertase (C3bBb) and factor I-generated degradation products (iC3b, C3c, C3dg)
CLINICAL ISSUES
Epidemiology
Incidence
Rare; estimated at 1-3 cases/million
Familial cases even rarer (˜ 6 patients reported)
Age
Primarily in children 5-15 years old
Increasingly appreciated in adults in recent series
˜ 55% are > 16 years old
40% of adults are > 60 years old
Gender
Female:male = 1.5:2
Presentation
Hematuria (˜ 90%)
Macrohematuria (˜ 15%)/nephritic presentation
Proteinuria (˜ 95%)
Nephrotic range (˜ 60%)
Proteinuria may undergo rapid fluctuation
Renal insufficiency (˜ 50%)
Acquired partial lipodystrophy (APL) (3-5%)
Loss of subcutaneous fat in upper 1/2 of body may precede kidney disease onset by several years
˜ 83% of APL patients have low C3 levels and polyclonal C3NeF
˜ 20% go on to develop MPGN after median of 8 years after onset of lipodystrophy
Ocular drusen common
˜ 10% develop decreased visual acuity
Laboratory Tests
Decreased serum C3 in ˜ 80% of patients
More common in children (100%) than adults (40%)
Increased C3dg and C3d
C3NeF (antibody to C3bBb) present in > 80% of MPGN II patients
Persists in > 50% during disease course
C3NeF present in ˜ 50% of MPGN, type I or III
CFH mutations
Treatment
Drugs
Steroids, immunosuppression not effective
Complement inhibition with eculizumab (anti-CD5 antibody) under study
Heparinoids may be used to protect GBM from complement activation
Plasmapheresis and plasma exchange in patients with CFH mutations
Recombinant CFH
Prognosis
Spontaneous remission rare
˜ 50% develop ESRD within 10-15 years
Mean time to ESRD is 5 years in adults, 20 years in children
Recurs in almost all renal allografts
˜ 50% of allografts ultimately fail, typically in 1st 3 years
MICROSCOPIC PATHOLOGY
Histologic Features
Glomerular patterns
Membranoproliferative glomerulonephritis (25-45%)
Mesangial proliferation, thickened GBM, duplication sometimes evident
Eosinophilic and refractile and stain brightly with PAS; fuchsinophilic on trichrome
DDD deposits stain poorly with Jones stain
Mild mesangial hypercellularity (30-50%)
Normal GBM by light microscopy
Crescentic glomerulonephritis (10-20%)
Focal crescents in > 50% of cases
Acute glomerulonephritis (10-20%)
Poly- or mononuclear cells, mesangial hypercellularity, normal thickness of GBM
Necrosis uncommon (˜ 15%)
Focal, segmental, and global glomerulosclerosis, late
Tubules and interstitium
Usually not affected early in disease
Thickened TBM sometimes evident due to deposits
Tubular atrophy and intersitial fibrosis develop late in disease
Vessels
No specific changes
Follow-up biopsies
Less acute glomerular inflammation
Increased glomerulosclerosis, tubular atrophy, fibrosis
Transitions
Progression from mesangial proliferative GN to MPGN pattern
Resolution of crescentic GN to mesangial proliferative GN



