| Disorder | Pathology |
|---|---|
| Alzheimer’s disease | Amyloid/tau pathology |
| Dementia with Lewy bodies Parkinson’s dementia Multisystem atrophy | Alpha-synuclein pathology |
| Frontotemporal dementia Progressive supranuclear palsy Corticobasilar degeneration | Tau pathology |
| Huntington’s disease Spinocerebellar ataxia | Trinucleotide repeat |
| Wilson’s disease (copper) Hallervorden–Spatz disease (iron) | Toxic/metabolic |
| Metachromatic leukodystrophy | Leukodystrophy |
| Creutzfeldt–Jakob disease Variant Creutzfeldt–Jakob disease (bovine spongiform encephalopathy) Gerstmann–Sträussler–Scheinker disease Fatal familiar insomnia (thalamic dementia) | Prion-related dementias |
Just because a patient develops memory disturbance does not mean it is Alzheimer’s disease (Table 13-2). Alzheimer’s dementia is perhaps the best-known and commonest dementia, but it is often the other symptoms associated with memory loss that help make the diagnosis clinically (Table 13-2). Just to complicate things, many patients have mixed types of dementia, particularly Alzheimer’s dementia plus dementia with Lewy bodies, or Alzheimer’s dementia plus vascular dementia (Figure 13-1). Such cases are complicated to diagnose clinically, and definitive diagnosis sometimes must await autopsy. Most dementias are really pathological diagnoses, not clinical diagnoses.
| Dementia | Clinical features |
|---|---|
| Alzheimer’s disease | Memory deficit |
| Aphasia | |
| Apraxia | |
| Agnosia | |
| Dementia with Lewy bodies | Memory deficit |
| Fluctuating attention | |
| Extrapyramidal signs | |
| Psychosis (hallucinations) | |
| Frontotemporal dementia | Memory deficit |
| Speech/language disorders | |
| Disinhibition | |
| Hyperorality | |
| Huntington’s disease | Memory deficit |
| Executive dysfunction | |
| Chorea | |
| Creutzfeldt–Jakob disease | Memory deficit |
| Ataxia | |
| Myoclonus | |
| Language disturbance |
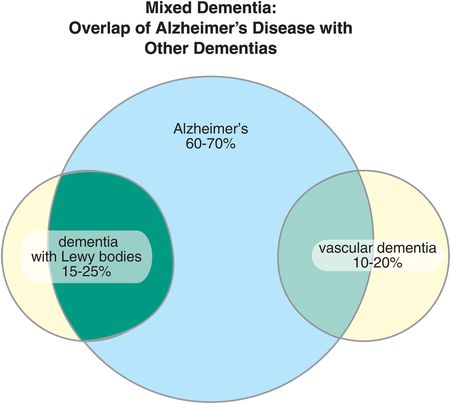
Figure 13-1. Mixed dementia. There are several types of dementia, of which Alzheimer’s disease is the most common. They are distinguished by their underlying pathologies. It is possible to have more than one dementia, and in fact many patients have both Alzheimer’s disease and either dementia with Lewy bodies or vascular dementia.
A wide variety of dementias are considered nondegenerative, and these are listed in Table 13-3. Many of these are treatable upon discovering the underlying cause, but others are not. Extensive clinical evaluation and laboratory testing must rule out these causes prior to concluding that a case of dementia is due to Alzheimer’s disease.
| Vascular | Multi-infarct dementia Strategic single-infarct dementia Small vessel disease Watershed area hypoperfusion |
| Infectious | HIV dementia Neurosyphilis Whipple’s disease Progressive multifocal leukoencephalopathy Tuberculosis Fungal/protozoal Sarcoidosis |
| Demyelinating | Multiple sclerosis |
| Endocrine | Hypothyroidism Cushing’s syndrome Adrenal insufficiency Hypoparathyroidism Hyperparathyroidism |
| Brain injuries | Post-anoxic Post-encephalitic Chronic subdural hematoma |
| Vitamin deficiency | B12, B1, folate, niacin |
| Vasculitides | Lupus erythematosus Sjögren’s disease |
| Toxicities | Heavy metal (storage) disorders (arsenic, mercury, lead) Industrial/environmental toxins (fertilizers, pesticides) Medications Chronic alcohol/drug abuse Wernicke–Korsakoff syndrome Marchiafava–Bignami disease |
| Organ failure | Hepatic encephalopathy Uremic encephalopathy Pulmonary insufficiency |
| Other causes | Dementia syndrome of depression Normal pressure hydrocephalus Nonconvulsive status epilepticus Acute intermittent poyphyria |
Alzheimer’s disease: β-amyloid plaques and neurofibrillary tangles
Without the introduction of disease-modifying treatments, Alzheimer’s disease is poised for an exponential increase throughout the world, with projections that it will quadruple over the next 40 years to affect 1 in every 85 people on earth: over 100 million people by 2050. Fortunately, new treatments are being designed to interfere with various known pathological processes, particularly the formation of amyloid plaques, in an attempt to halt or slow disease progression in Alzheimer’s disease before neurons are irretrievably lost. To understand the current diagnostic criteria for Alzheimer’s disease, how and why biomarkers are being integrated into the diagnosis of this disorder, and the rationale behind the hot pursuit of new therapeutics, it is necessary to understand how the two hallmarks of this disorder, amyloid plaques and neurofibrillary tangles, are thought to be formed in the brain in Alzheimer’s disease.
The amyloid cascade hypothesis
The leading contemporary theory for the biological basis of Alzheimer’s disease centers around the formation of toxic amyloid plaques from peptides due to the abnormal processing of amyloid precursor protein (APP) into toxic forms of Abeta (Aβ) peptides (Figures 13-2 through 13-9). Why do we make Aβ in the first place? Although this is not fully understood, nontoxic Aβ peptides have antioxidant properties, can chelate metal ions, regulate cholesterol transport, and may be involved in blood vessel repair, as a sealant at sites of injury or leakage, possibly protecting from acute brain injury. Hypothetically, Alzheimer’s disease is a disorder in which toxic Aβ peptides are formed, leading to deposition of amyloid plaque in the brain, with the ultimate destruction of neurons diffusely throughout the brain, somewhat analogous to how the abnormal deposition of cholesterol in blood vessels causes atherosclerosis.
Thus, Alzheimer’s disease may be essentially a problem of too much formation of Aβ amyloid-forming peptides, or too little removal of them. One idea is that neurons in some patients destined to have Alzheimer’s disease have abnormalities either in genes that code for a protein called amyloid precursor protein (APP), or in the enzymes that cut this precursor into smaller peptides, or in the mechanisms of removal of these peptides from the brain and from the body. APP is a transmembrane protein with the C-terminal inside the neuron and the N-terminal outside the neuron. One pathway for APP processing does not produce toxic peptides and involves the enzyme α-secretase (Figure 13-2). Alpha-secretase cuts APP close to the area where the protein comes out of the membrane, forming two peptides: a soluble fragment known as α-APP and a smaller 83-amino-acid peptide that remains embedded in the membrane until it is further cleaved by a second enzyme acting within the neuronal membrane, called γ-secretase (Figure 13-2). That enzyme produces two smaller peptides, p7 and p3, which are apparently not “amyloidogenic” and therefore not toxic (Figure 13-2).
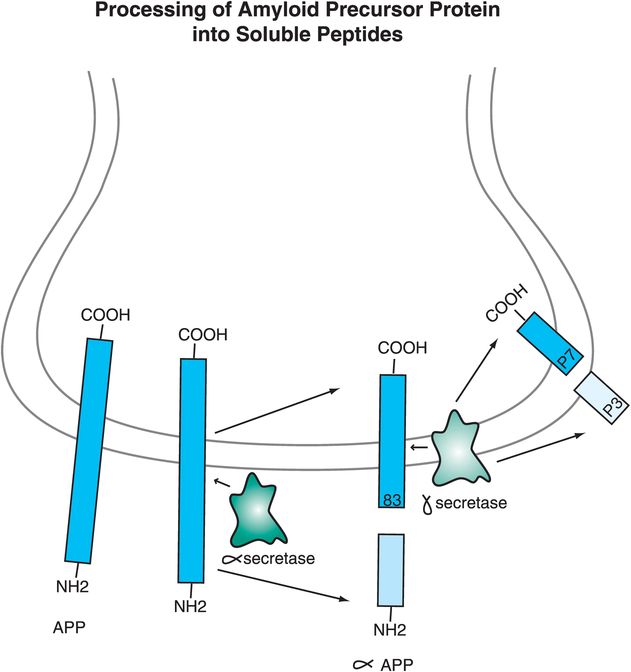
Figure 13-2. Processing of amyloid precursor protein into soluble peptides. The way in which amyloid precursor protein (APP) is processed may help determine whether an individual develops Alzheimer’s disease or not. A nontoxic pathway for APP processing is shown here. APP is a transmembrane protein with the C-terminal inside the neuron and the N-terminal outside the neuron. The enzyme α-secretase cuts APP close to where it comes out of the membrane to form two peptides: α-APP, which is soluble, and an 83-amino-acid peptide that remains in the membrane. A second enzyme, γ-secretase, cuts the embedded peptide into two smaller peptides, p7 and p3, which are not “amyloidogenic” and thus are not toxic.
Another pathway for APP processing can produce toxic peptides that form amyloid plaques (i.e., “amyloidogenic” peptides). In this case a different enzyme, β-secretase, cuts APP a little bit further away from the area where APP comes out of the membrane, forming two peptides: a soluble fragment known as β-APP and a smaller 91-amino-acid peptide that remains embedded in the membrane until it is further cleaved by γ-secretase within the membrane (Figure 13-3). This releases Aβ peptides of 40, 42 or 43 amino acids that are “amyloidogenic,” especially Aβ42 (Figure 13-3).
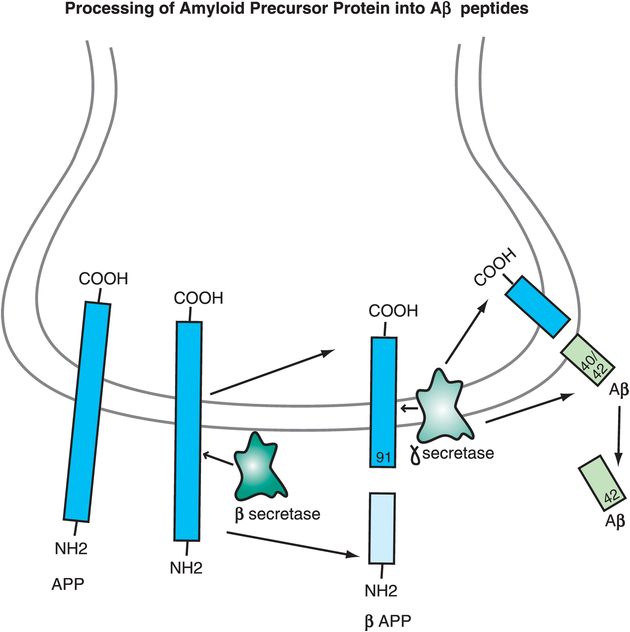
Figure 13-3. Processing of amyloid precursor protein into Aβ peptides. The way in which amyloid precursor protein (APP) is processed may help determine whether an individual develops Alzheimer’s disease or not. A toxic pathway for APP processing is shown here. APP is a transmembrane protein with the C-terminal inside the neuron and the N-terminal outside the neuron. The enzyme β-secretase cuts APP at a spot outside the membrane to form two peptides: β-APP, which is soluble, and a 91-amino-acid peptide that remains in the membrane. Gamma-secretase then cuts the embedded peptide; this releases Aβ peptides of 40, 42, or 43 amino acids. These toxic (amyloidogenic) peptides form amyloid plaques.
In Alzheimer’s disease, genetic abnormalities may produce an altered APP that, when processed by this second pathway involving β-secretase, produces smaller peptides that are especially toxic. Individuals who do not get Alzheimer’s disease may produce peptides that are not very toxic, or may have highly efficient removal mechanisms that prevent neuronal toxicity from developing. The amyloid cascade hypothesis of Alzheimer’s disease therefore begins with an APP that is hypothetically genetically abnormal, or genetically or environmentally abnormal in the way it is processed, so that when it is cut into smaller peptide fragments too many toxic peptides are made, accumulate, and form neuron-destroying amyloid plaques, i.e., amyloidosis, and neurofibrillary tangles. Hypothetically, this process triggers a lethal chemical cascade that ultimately results in Alzheimer’s disease (Figures 13-3 through 13-8).
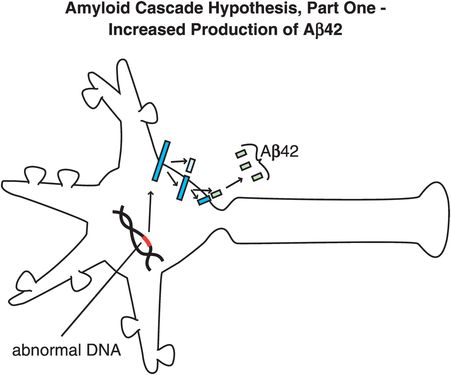
Figure 13-4. Amyloid cascade hypothesis, part 1: increased production of Aβ42. One theory for the pathophysiology of Alzheimer’s disease is that there are genetic abnormalities in amyloid precursor protein (APP), so that when it is processed by the pathway involving β-secretase, it produces smaller, toxic peptides (especially Aβ42, as shown here).
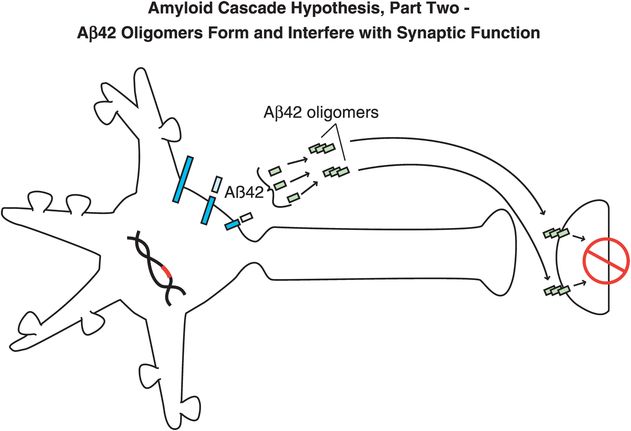
Figure 13-5. Amyloid cascade hypothesis, part 2: Aβ42 oligomers form and interfere with synaptic function. Aβ42 peptides assemble together to form oligomers, which interfere with synaptic functioning and neurotransmitter actions but are not necessarily lethal to neurons.
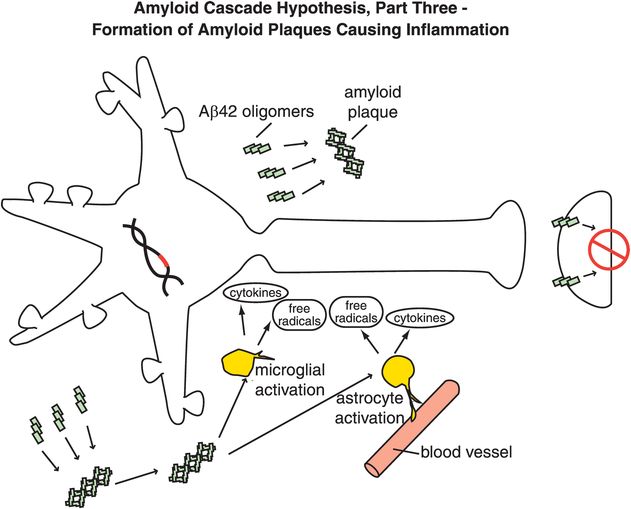
Figure 13-6. Amyloid cascade hypothesis, part 3: formation of amyloid plaques causing inflammation. Aβ42 oligomers clump together along with other molecules to form amyloid plaques. These plaques can cause inflammatory responses, activation of microglia and astrocytes, and release of toxic chemicals such as cytokines and free radicals.
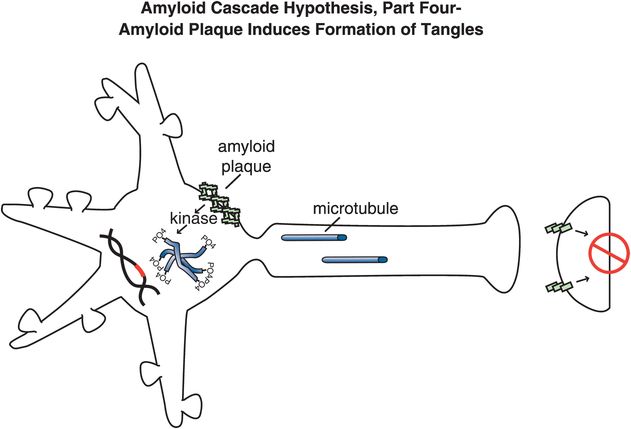
Figure 13-7. Amyloid cascade hypothesis, part 4: amyloid plaque induces formation of tangles. Amyloid plaques and the chemical events they cause activate kinases, cause phosphorylation of tau proteins, and convert microtubules into tangles within neurons.
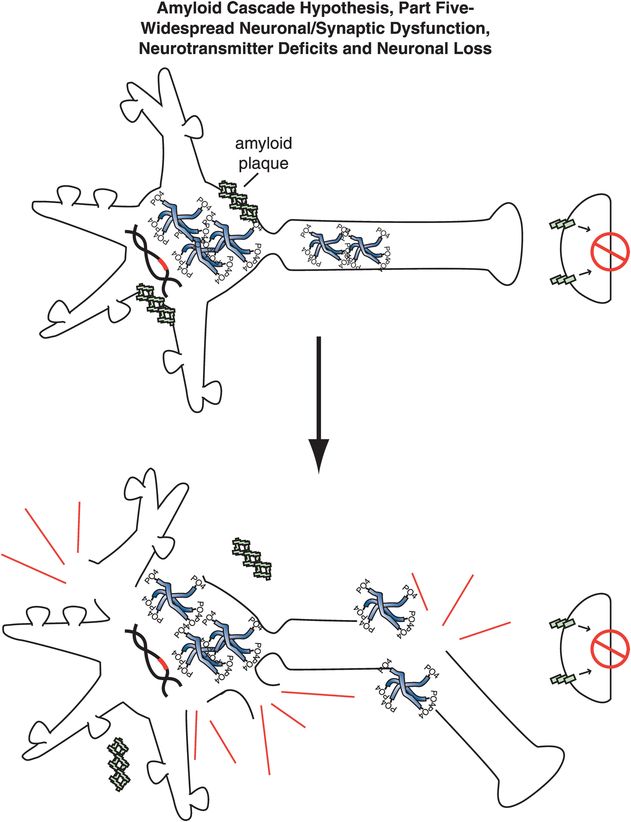
Figure 13-8. Amyloid cascade hypothesis, part 5: neuronal dysfunction and loss. The effects of amyloid plaques and the build-up of neurofibrillary tangles can ultimately lead to neuronal dysfunction and death.
Specifically, abnormal genes or other influences cause the formation of an altered APP, or altered processing into too many toxic Aβ42 peptides (Figure 13-4). Next, the Aβ42 peptides form oligomers (a collection of a few copies of Aβ42 assembled together: Figure 13-5). These oligomers can interfere with synaptic functioning and neurotransmitter actions such as those of acetylcholine, but they are not necessarily lethal to the neurons at first. Eventually, Aβ42 oligomers form amyloid plaques, which are even larger clumps of Aβ42 peptides stuck together with a number of other molecules (Figure 13-6). A number of nasty biochemical events then occur, including inflammatory responses, activation of microglia and astrocytes, and release of toxic chemicals including cytokines and free radicals (Figure 13-6). These chemical events then hypothetically trigger the formation of neurofibrillary tangles within neurons by altering the activities of various kinases and phosphatases, causing hyperphosphorylation of tau proteins, and converting neuronal microtubules into tangles (Figure 13-7). Finally, widespread synaptic dysfunction from Aβ42 oligomers, neuronal dysfunction and death from formation of amyloid plaques outside of neurons and neurofibrillary tangles within neurons leads to diffuse neuronal death (Figure 13-8) and regional expansion of neuronal destruction in the cortex, causing the relentless progression of Alzheimer’s symptoms of amnesia, aphasia, agnosia, apraxia, and executive dysfunction. Some investigators believe that Alzheimer’s disease may spread from neuron to neuron, with pathological phosphorylated tau transported down axons, released at synapses and then taken up by neighboring cells. Pathological tau possibly then latches onto normal tau in the connected neurons, triggering the formation of new pathological mis-folded tau, from one affected neuron to the next.
Support for the amyloid cascade hypothesis comes from genetic studies of those relatively rare inherited autosomal dominant forms of Alzheimer’s disease. Sporadic (i.e., noninherited) cases account for the vast majority of Alzheimer’s disease cases, but inherited cases can provide clues for what is wrong in the usual sporadic cases of Alzheimer’s disease. Rare familial cases of Alzheimer’s disease have an early onset (i.e., before age 65) and have been linked to mutations in at least three different chromosomes: 21, 14, and 1. The mutation on chromosome 21 codes for a defect in APP, leading to increased deposition of β-amyloid. Recall that Down’s syndrome is also a disorder of this same chromosome (i.e., trisomy 21), and virtually all such persons develop Alzheimer’s disease if they live past age 50. A different mutation on chromosome 14 codes for an altered form of a protein called presenilin 1, a component of the γ-secretase enzyme complex. A third mutation, on chromosome 1, codes for an altered form of presenilin 2, a component of a different form of γ-secretase. It is not yet clear what if anything these three mutations in the rare familial cases tell us about the pathophysiology of the usual sporadic, nonfamilial, and late-onset cases of Alzheimer’s disease. However, they all point to abnormal processing of APP into amyloidogenic β-amyloid peptides as a cause for the dementia, consistent with the amyloid cascade hypothesis. Theoretically, different abnormalities in amyloid processing may occur in sporadic Alzheimer’s disease from those identified in inherited cases, and there may even be multiple abnormalities that could be responsible for sporadic Alzheimer’s disease as a final common pathway, but the evidence nevertheless implicates something in the amyloid cascade that goes wrong in Alzheimer’s disease. If so, this implies that preventing the formation of amyloidogenic peptides could prevent Alzheimer’s disease.
ApoE and risk of Alzheimer’s disease
A corollary to the amyloid cascade hypothesis is the possibility that something may be wrong with a protein that binds to amyloid peptides in order to remove them (Figure 13-9). This protein is called apolipoprotein E (ApoE). In the case of “good” ApoE, it binds to β-amyloid peptides and removes them, hypothetically preventing the formation of Alzheimer’s disease and dementia (Figure 13-9A). In the case of “bad” ApoE, a genetic abnormality in the formation of ApoE causes it to be ineffective in how it binds to β-amyloid peptides. This causes amyloid plaques to be formed and deposited around neurons, which goes on to damage neurons and cause Alzheimer’s disease (Figure 13-9B).
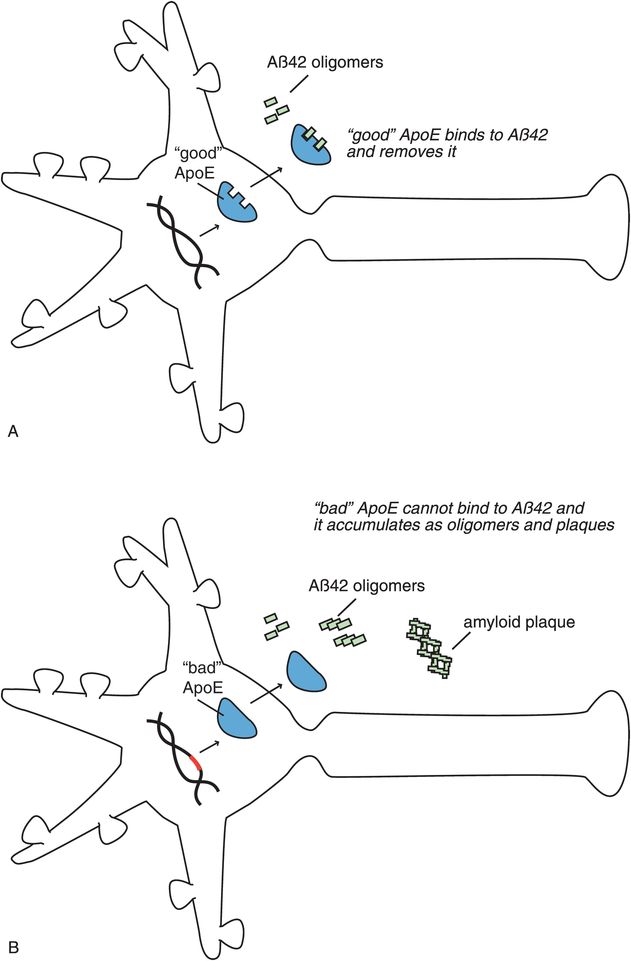
Figure 13-9. ApoE and Alzheimer’s disease. Another version of the amyloid cascade hypothesis is the possibility that something is wrong with the protein apolipoprotein E (ApoE). (A) Properly functioning (“good”) ApoE binds to β-amyloid and removes it, thus preventing development of Alzheimer’s disease and dementia. (B) An abnormality in DNA could lead to the formation of a defective or “bad” version of the ApoE protein, such that it cannot effectively bind to amyloid. This would prevent removal of amyloid, allowing it to accumulate and damage neurons, so that Alzheimer’s disease develops.
Genes coding for ApoE are associated with different risks for Alzheimer’s disease. There are three alleles (or variants) of this gene coding for this apolipoprotein called E2, E3, and E4, and everyone has two alleles. The E4 variant on chromosome 19 (“bad” ApoE) is linked to many cases of late-onset Alzheimer’s disease, the usual form of this illness. ApoE is associated with cholesterol transport and involved with other neuronal functions including repair, growth, and maintenance of myelin sheaths and cell membranes. Having one or two copies of E4 increases the risk of getting Alzheimer’s disease. In fact, some studies show that you have a 50–90% chance of developing Alzheimer’s disease by age 85 if you are an E4 homozygote (i.e., you have two copies of E4); a 45% chance if you are a heterozygote for E4, versus the risk in the general population at 20%. Alzheimer’s patients with the E4 gene also have more amyloid deposits and progress more rapidly to dementia than those without the E4 gene. The E2 variant may actually be somewhat protective.
Three stages of Alzheimer’s disease
The old way of diagnosing Alzheimer’s dementia was neurological and neuropsychological testing for the tentative clinical diagnosis of possible or probable Alzheimer’s disease and then postmortem evaluation for confirmation of an actual diagnosis of Alzheimer’s disease. In 2011, the diagnostic criteria were revised in two major ways: firstly, they expanded the notion of Alzheimer’s disease into three stages to reflect the current dynamic sequence model of structural and functional brain changes over time in elderly people who are first cognitively normal, then have mild cognitive changes, and finally develop Alzheimer’s disease (Figure 13-10). Secondly, the new diagnostic criteria have incorporated biomarkers. The five biomarkers in the new Alzheimer’s dementia criteria include both biomarkers of amyloidosis/amyloid accumulation and biomarkers of neurodegeneration (Table 13-4).
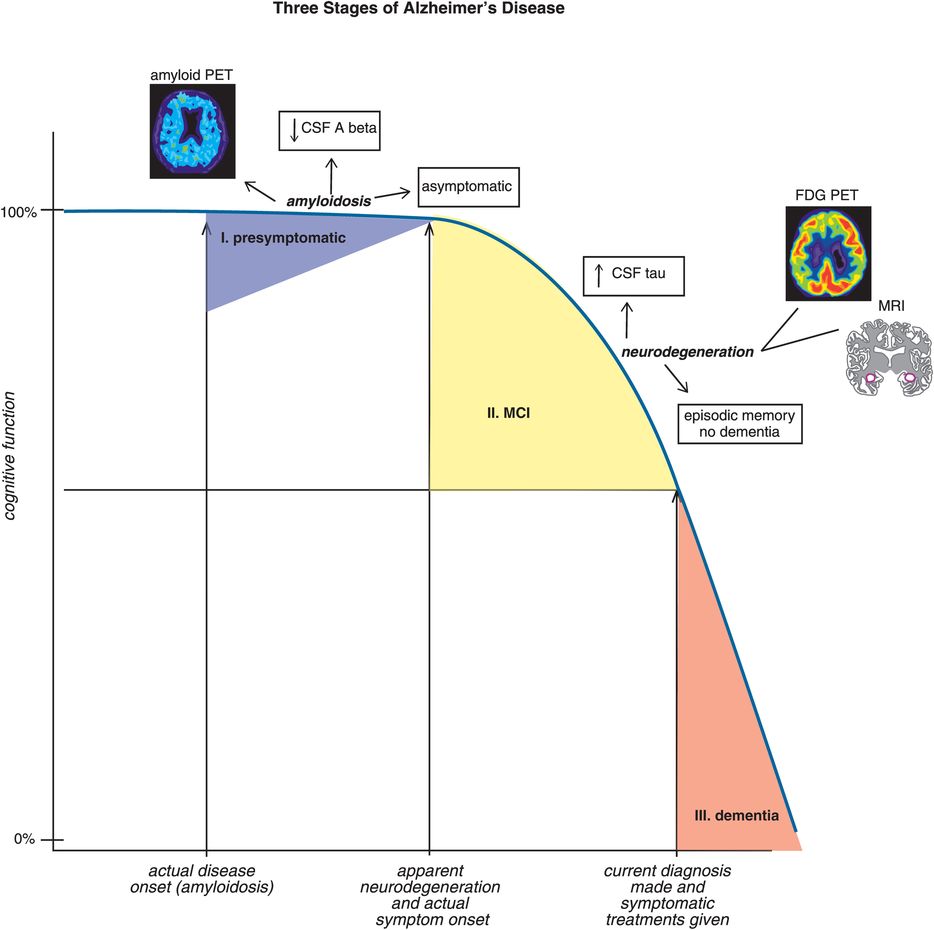
Figure 13-10. The three stages of Alzheimer’s disease. During stage I of Alzheimer’s disease (presymptomatic), cognition is intact despite elevated levels of brain amyloid as evidenced by both positive amyloid positron emission tomography (PET) and reduced levels of Aβ toxic peptides in cerebrospinal fluid (CSF). Clinical signs of cognitive impairment in the form of episodic memory deficits begin to manifest during stage II (mild cognitive impairment, MCI). The onset of clinical symptoms in stage II appears to be correlated with neurodegeneration, as evidenced by elevated CSF tau, brain glucose hypometabolism on fluorodeoxyglucose PET (FDG-PET) scans and volume loss in key brain regions on magnetic resonance imaging (MRI) scans. During stage III of Alzheimer’s disease (dementia), cognitive deficits can be severe. Currently, treatment of Alzheimer’s symptoms does not typically begin until stage III, long after the actual disease onset.
| Biomarkers of amyloid accumulation | Biomarkers of neurodegeneration |
|---|---|
| Abnormal radioactive tracer retention on amyloid PET scans | Elevated CSF tau (total and phosphorylated tau) |
| Low CSF amyloid levels of Aβ42 | Decreased FDG uptake on PET |
Atrophy on structural magnetic resonance imaging
|
CSF, cerebrospinal fluid; FDG, fluorodeoxyglucose; PET, positron emission tomography.
First stage of Alzheimer’s disease: preclinical (asymptomatic amyloidosis)
It seems obvious that the dementia of Alzheimer’s disease does not occur as soon as the first amyloid plaque arrives in the brain. Early plaques in fact seem to be relatively asymptomatic, but somewhere along the way, sufficient accumulation of them seems to trigger the neurodegeneration, or at least is associated with the neurodegeneration, that leads to dementia. It is not clear whether amyloid is an epiphenomenon of the neurodegenerative process, or whether amyloid drives the neurodegenerative process. Amyloid biomarkers are certainly assisting in the diagnostic process for identifying early stages of Alzheimer’s disease. Therapeutics, discussed below, includes many agents in clinical testing that interfere with amyloid accumulation, hypothesizing that amyloid drives neurodegeneration, and that interfering with amyloidosis will halt or delay the progress of Alzheimer’s disease. However, the lack of convincing evidence of clinical benefit of this approach to date suggests that another process may be the culprit causing neurodegeneration while amyloid is accumulating.
Here we will discuss how amyloid imaging is enhancing the diagnostic accuracy of Alzheimer’s disease in its early stages. The first stage of Alzheimer’s disease is now considered to be preclinical and silent, but trouble is brewing (Figures 13-10 and 13-11). That trouble is the slow, relentless deposition of Aβ peptides into the brain rather than their elimination via the CSF, plasma, and liver. This presymptomatic stage can now be identified with biomarkers (Table 13-4; Figures 13-10 and 13-11): for example, CSF levels of Aβ are low because Aβ is being deposited in the brain instead of leaving the brain. Furthermore, amyloidosis is detectable with PET scans at the presymptomatic stage using radioactive neuroimaging tracers that label amyloid plaques (Figure 13-11). Tracers bind to the fibrillar form of amyloid and thus label mature neuritic plaques which can be seen on PET scans after administering a radioactive chemical that binds to amyloid.
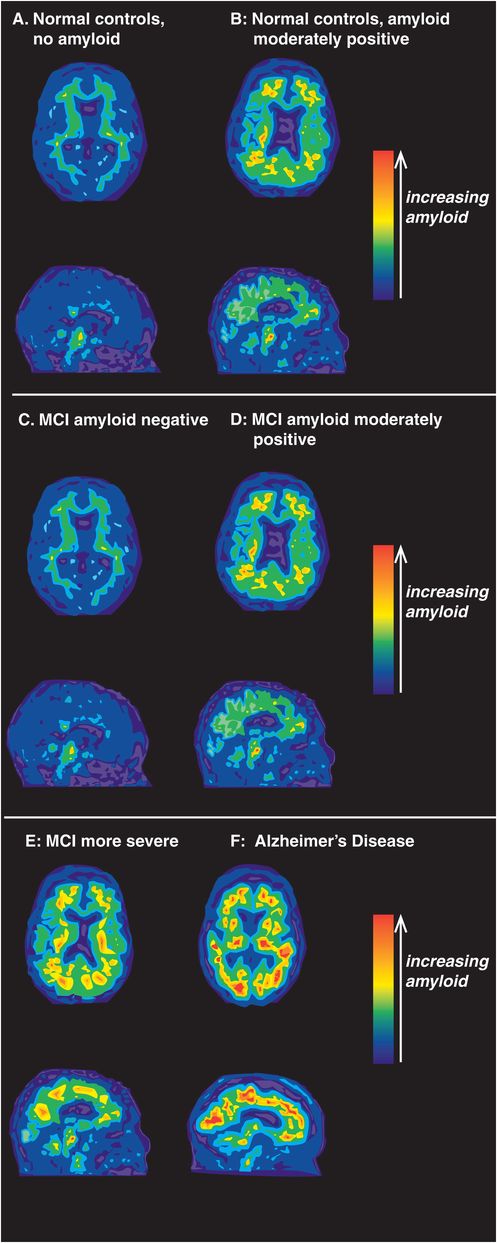
Figure 13-11. Amyloid PET imaging. Positron emission tomography (PET) using amyloid tracers can be used to detect the presence of amyloid during the progression of Alzheimer’s disease. In cognitively normal controls (A), amyloid PET imaging shows the absence of amyloid. Individuals who are cognitively normal but have moderate accumulation of amyloid (B) are likely in the presymptomatic first stage of Alzheimer’s disease. Although mild cognitive impairment (MCI) is often present in the prodromal second stage of Alzheimer’s disease, not all patients with MCI have brain amyloid deposition (C). In such cases, the clinical presence of cognitive impairments is likely attributable to a cause other than Alzheimer’s disease. Unfortunately, MCI is often a harbinger of impending Alzheimer’s dementia. In these cases (D), amyloid deposition accompanies cognitive impairments and both amyloid accumulation and clinical symptoms of MCI worsen as Alzheimer’s disease progresses (E). In the third and final stage of Alzheimer’s disease, when full-blown dementia is clinically evident, a large accumulation of brain amyloid can readily be seen (F).
Interestingly, amyloid is rarely detected in the brains of individuals under the age of 50, even those with the high-risk E4 genotype. Although most cognitively normal healthy elderly people show no evidence of amyloid deposition (13-11A), about a quarter of cognitively normal elderly controls are amyloid positive (Figure 13-11B), and are thus considered to have presymptomatic Alzheimer’s disease (Figure 13-10). About half of patients with MCI show no evidence of amyloid deposition (Figure 13-11C), but the other half do show either moderate (Figure 13-11D) or severe amyloid deposition (Figure 13-11E). Almost 100% of patients with clinically probable Alzheimer’s disease show heavy amyloid deposition (Figure 13-11F).
Thus, Aβ amyloid pathology is not specific for the dementia phase of Alzheimer’s disease, but may mean that the fuse is already lit in the presymptomatic phase. Serial amyloid scans show an annual increase of up to 4% of amyloid in patients with probable Alzheimer’s disease. Although amyloid can be seen in the presymptomatic stage of Alzheimer’s disease, by definition, clinical changes are not detectable at this stage, presumably because there is not much neurodegeneration yet. It is not the amyloid plaques per se, but the neurodegeneration with which they are later associated, that seems to correlate with the onset of symptoms in the second and third stages of Alzheimer’s disease.
Most worrisome for the eventual progression of presymptomatic Alzheimer’s disease to the MCI symptomatic stage of illness is that some studies suggest that Aβ deposition in the preclinical stage is already associated with some degree of gray-matter atrophy in the hippocampus and the posterior cingulate gyrus that can be demonstrated with structural MRI scanning (Figure 13-12). Cognitively normal elderly adults with the E4 genotype have greater hippocampus volume loss than do cognitively normal adults without E4. Furthermore, those cognitively normal elderly adults with the E4 genotype exhibit faster atrophy, so both elevated Aβ levels and the E4 genotype are associated with gray-matter atrophy in subjects even without cognitive impairment. Reliable atrophy and cortical thinning are identifiable in the hippocampus and in entorhinal, temporal, and parietal cortices in asymptomatic individuals nearly a decade before the onset of dementia.
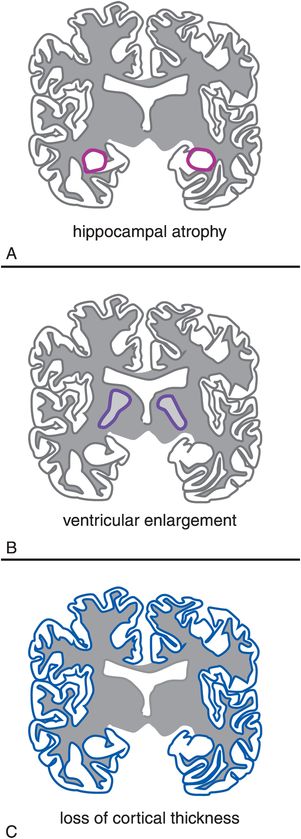
Figure 13-12. Structural MRI. Although hippocampal atrophy (A) and ventricular enlargement (B) are seen with normal aging, the progression of this volume loss is significantly more rapid in patients with Alzheimer’s disease. The brains of patients with Alzheimer’s disease also show a progressive thinning of the cortex (C).
Now that biomarkers are clarifying this first stage of presymptomatic Alzheimer’s disease, it has come to be viewed as the leading edge of a continuum of a process of formation of plaques and tangles causing a relentless march towards dementia. Since some patients may develop amyloid plaques but do not progress to neurodegeneration or dementia, the notion of a preclinical stage is intended for research purposes at this time in order to sort out more reliably who is destined to progress (and thus possibly who to treat at this stage with anti-amyloid therapies on the horizon) and who is not. Early brain changes at the preclinical stage that herald clinical progression to Alzheimer’s dementia is consistent with the current amyloid cascade hypothesis, but at this point it is not proven for whom this is true, nor how to apply an individual patient’s biomarker results to make an accurate prognosis for that individual. A major research question remains as to whether there is a threshold level of Aβ deposition at which brain atrophy occurs, or whether there is another process occurring later and then in parallel that causes the brain atrophy. The exact definition of pathological levels of Aβ deposition (Figure 13-11) remains open, which is why this presymptomatic stage of Alzheimer’s disease with amyloidosis but no symptoms is a research diagnosis now.
Risk factors at this presymptomatic stage of illness that may hasten the pace or increase the likelihood of progressing to dementia include depression, type 2 diabetes, ApoE4 genotype, and vascular disease, particularly cerebral emboli. Some experts even wonder whether the effects of type 2 diabetes in the brain can be called “type 3 diabetes,” since toxic amyloid peptides are overexpressed in the fat cells of obese individuals, and in the brains of Alzheimer’s patients with dementia insulin concentrations, insulin-like growth factor, and insulin receptors are decreased by up to 80%. Since insulin modulates neurotransmitter release, tubulin activity, neuronal survival, and synaptic plasticity, losing your insulin from diabetes does not look like a good idea if you want to prevent Alzheimer’s dementia. Thus factors that prevent type 2 diabetes may also help prevent the progression of preclinical Alzheimer’s disease to dementia, but that is still a matter of investigation – although it does seem like a matter of common sense. Other common-sense factors that may promote healthy brain aging but are not yet proven to slow the progression of preclinical Alzheimer’s disease to dementia include:
- a healthy diet
- adequate sleep
- daily exercise
- active, socially integrated lifestyle
- leisure activities
- cognitive stimulation
- optimized treatment of depression and other mental illnesses
- meditation and other mindfulness strategies (e.g., yoga)
- spiritual activities
- controlling vascular risk factors (hypertension, diabetes, dyslipidemia, obesity)
Second stage of Alzheimer’s disease: mild cognitive impairment (MCI) (symptomatic, predementia stage of amyloidosis plus some neurodegeneration)
Patients with mild cognitive impairment (MCI) have mild cognitive symptoms but not dementia. Some call this stage of disease “predementia Alzheimer’s disease,” or “MCI due to Alzheimer’s disease” or even “prodromal Alzheimer’s disease.” However, this diagnosis of MCI does not yet mean that Alzheimer’s disease pathology has necessarily caused the symptoms or even that MCI patients will inevitably progress to dementia. In fact, there is great debate about what is MCI versus what is “normal aging.” Hopefully, the study of biomarkers will be able to settle this in the future. From a purely clinical perspective, over half of elderly residents living in the community complain of memory impairment. They have four common complaints: compared to their functioning of 5–10 years ago, they experience diminished ability (1) to remember names, (2) to find the correct word, (3) to remember where objects are located, and (4) to concentrate. When such complaints occur in the absence of overt dementia, depression, anxiety disorder, sleep/wake disorder, pain disorder, or ADHD (attention deficit hyperactivity disorder) it is called MCI. As already mentioned, the new diagnostic criteria, coupled with biomarkers, are attempting to make the distinction among those with normal aging, those with reversible conditions, and those with MCI destined to progress to the dementia stage of Alzheimer’s disease. On clinical grounds alone and without biomarkers, studies show that between 6% and 15% of MCI patients convert to a diagnosis of dementia every year; after 5 years about half meet the criteria for dementia; after 10 years or autopsy, up to 80% will prove to have Alzheimer’s disease. Also, MCI patients who have depression exhibit more neurodegeneration and brain atrophy than MCI patients without depression.
Biomarker studies seek to determine who among these MCI patients are destined to progress inevitably to Alzheimer’s dementia and who are the lucky ones with a benign and nonprogressive condition. Already foreseen is the need to identify the high-risk group of MCI patients in order to be able to treat them to prevent dementia rather than to treat them as we are doing now, after the brain has already degenerated and the third stage of Alzheimer’s disease has been reached. Also, there is the need to identify those with benign conditions or conditions other than those linked to neurodegeneration associated with amyloidosis, so that they are not needlessly treated with amyloid-targeted therapies which may be both expensive and have side effects.
Only a subset of MCI patients display measurable amyloidosis (Figure 13-11C, D, and E) and only a small proportion (about 10%) of MCI patients without amyloid progress to dementia. However, in those MCI patients with both cognitive symptoms and amyloidosis, the assumption is that they have progressed beyond a presumed earlier state of presymptomatic and silent amyloidosis without damage to the brain (Table 13-4: positive amyloid PET scans and low CSF Aβ levels) to early neurodegeneration (Table 13-4: high CSF tau levels plus structural neuroimaging abnormalities) (Figure 13-10). About half of amyloid-positive MCI patients progress to dementia within a year, and 80% may progress to dementia within 3 years, with MCI patients having the E4 genotype progressing even more rapidly. Those MCI patients who convert to dementia have higher amyloid loads, and the utility of amyloid PET to identify Alzheimer’s pathology in the setting of clinical MCI is becoming increasingly convincing. Impairment in episodic memory (the ability to learn and retain new information) is the cognitive symptom most commonly seen in those MCI patients who eventually progress to Alzheimer’s dementia. Those MCI patients with the E4 genotype have acceleration of the time to progression from MCI to dementia, so that amyloid imaging, plus testing of episodic memory, plus genetic determination of E4 may currently be the most valuable way to predict higher risk of progression to dementia from the MCI stage.
However, as already mentioned, brain amyloidosis alone does not appear sufficient to produce cognitive decline. Rather, neurodegeneration must occur as the probable direct substrate of cognitive impairment, with the rate of cognitive decline being driven by the rate of neurodegeneration, not by the rate of amyloid deposition. Neurodegenerative atrophy on structural magnetic resonance imaging (MRI) scans both precedes and parallels cognitive decline. The new diagnostic criteria for Alzheimer’s disease suggest that it is important to determine whether an MCI patient with impairment of episodic memory has neurodegeneration (as well as amyloidosis and the E4 genotype), to help distinguish MCI that is destined to progress to dementia from nonprogressive MCI and normal aging. Thus, the MCI stage of Alzheimer’s disease by definition not only has amyloidosis and cognitive symptoms, but also biomarker evidence of neurodegeneration (Table 13-4; Figures 13-10 through 13-13).
One biomarker for neurodegeneration is the presence of elevated cerebrospinal fluid (CSF) tau (including phospho-tau), thought to be associated with neuronal loss in the brains of Alzheimer’s disease patients because it is also elevated in other neurodegenerative diseases such as stroke and Creutzfeldt–Jakob disease (Table 13-4). Numerous neuroimaging biomarkers of neurodegeneration also are available, from single photon emission computerized tomography (SPECT) and fluorodeoxyglucose positron emission tomography (FDG-PET) scans (Figure 13-13), to structural magnetic resonance imaging (MRI) of hippocampal atrophy and of cortical thinning (Figure 13-12), to functional MRI and beyond.
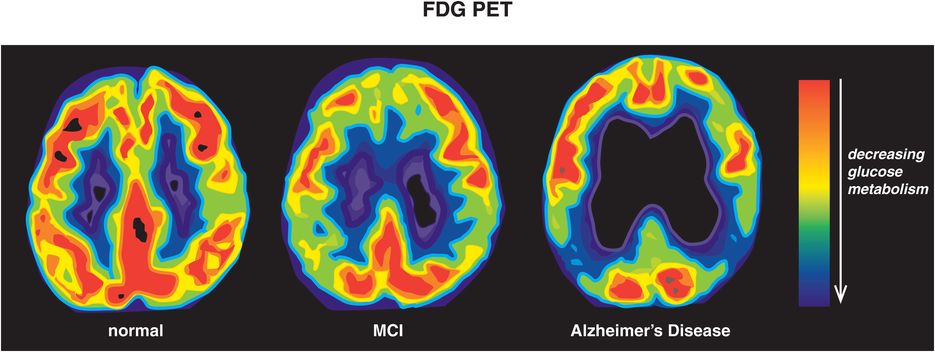
Figure 13-13. Glucose hypometabolism in Alzheimer’s disease. The brains of normal, healthy controls show robust glucose metabolism throughout the brain using fluorodeoxyglucose positron emission tomography (FDG-PET). During the early, prodromal stage of Alzheimer’s disease, when mild cognitive impairment (MCI) is present, there is a reduction in brain glucose metabolism in more posterior brain regions such as temporoparietal cortex. As the disease progresses to full-blown Alzheimer’s dementia, brain glucose hypometabolism becomes evident on FDG-PET. The worsening of glucose metabolism with the progression of Alzheimer’s disease is believed to reflect accumulating neurodegeneration, especially in key brain areas such as temporoparietal cortices.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


