I. INTRODUCTION. The three significant milestones in the history of clinical cytogenetics are the preparation of chromosome spreads from peripheral blood cultures (Exp Cell Res. 1960;20:613), the development of hypotonic methods to obtain enhanced chromosome spreads (Cancer Res. 1960;20:462), and the discovery that fluorescent quinacrine compounds could be used to demonstrate a unique banding pattern for each human chromosome pair (Hereditas. 1971;67:89). The remarkable advancement of the field of human cytogenetics is emphasized by the fact that it has been only 50 years since the correct number of human chromosomes was established. The various banding methods in current use not only permit identification of each chromosome, but also make it possible to detect specific alterations associated with hereditary syndromes and neoplasms.
II. TRADITIONAL CYTOGENETIC ANALYSIS. While cytogenetic analysis is commonly used in the evaluation of congenital disorders (specifically, to diagnose syndromes associated with abnormalities of chromosomal number or structure, to establish the chromosomal sex in cases of sexual ambiguity, and to screen for karyotypic abnormalities in patients with multiple birth defects) and for prenatal diagnosis, the technique’s primary application in surgical pathology is in the evaluation of neoplastic disorders. The utility of the technique in surgical pathology rests on the fact that specific cytogenetic abnormalities have been recognized that are closely, and sometimes uniquely, associated with morphologically and clinically distinct subsets of lymphoma and leukemia, or with soft tissue neoplasms. Cancer cytogenetic studies have greatly aided targeted therapy, prognosis, and risk-based stratification of intensity of therapy.
A. Advantages. The power of conventional cytogenetics lies in its ability to provide simultaneous analysis of the entire genome without any foreknowledge of the chromosomal regions involved in the disease process. In most cases, the type and location of an identified chromosomal abnormality is either directly diagnostic or can be used to direct additional testing. Contrary to some predictions, the advent of technologies such as array comparative genomic hybridization (aCGH) has not diminished the importance of traditional cytogenetics; in fact, these novel molecular techniques achieve some of their greatest utility when they are utilized in conjunction with traditional clinical cytogenetics.
B. Limitations. The clinical utility of traditional cytogenetic analysis is restricted by two general features of the method. From a technical standpoint, analysis can only be performed on viable tissue specimens that contain proliferating cells (discussed in more detail below). From a sensitivity standpoint, analysis has resolution of only about 3 to 4 Mb at an 850-band level, and only about 7 to 8 Mb at a 400-band level. Traditional cytogenetic analysis is therefore only suited for detection of numerical abnormalities and gross structural rearrangements. The method does not have the sensitivity to detect mutations such as small deletions and amplifications, single base pair substitutions, and so on.
C. Basic laboratory procedures. Chromosomes that can be individually distinguished by light microscopy can only be obtained during cell division, and so the fundamental requirement for traditional cytogenetic analysis is a tissue specimen that contains actively proliferating cells, or cells that can be induced to proliferate in vitro. The basic method for production of metaphase chromosomes for cytogenetic analysis is shown in Figure 58.1.
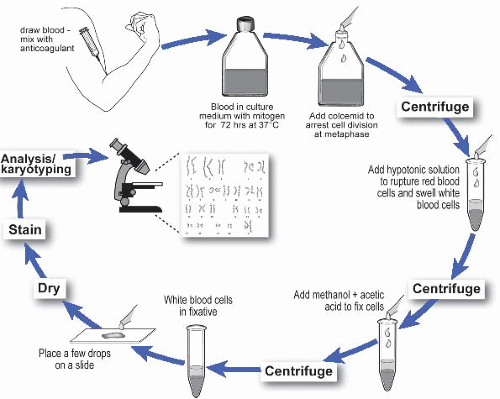
Figure 58.1 Overall scheme for the production of metaphase chromosomes for traditional cytogenetic analysis.
1. Culture initiation. Different specimen types have different sample and handling requirements (Table 58.1). Inappropriate handling, as well as delay between specimen collection and culture initiation, can markedly decrease the likelihood that the sample will grow in vitro, so communication and coordination with the cytogenetics laboratory are essential.
In vitro culture relies on a sterile microenvironment, and so specimens should be collected under sterile conditions. In practice, sterility is most difficult to achieve when sampling solid tissues; in this setting, clean instruments and a clean cutting surface, together with transport of the specimen in medium supplemented with broad-spectrum antibiotics, can be used to minimize contamination.
TABLE 58.1 Specimen Requirements
Tissue type
Sample collection
Peripheral blood
Preservative-free sodium heparin; transport refrigerated or at room temperature
Bone marrow aspirate
Preservative-free sodium heparin; the first several milliliters of the aspirate usually contains the greatest proportion of cells and so is the optimal sample for cytogenetic analysis; transport at room temperature
Solid tissue
Collect and transport in sterile culture medium containing broad-spectrum antibiotics; carefully select maximally viable tumor for analysis; transport on ice to minimize autolysis and microbial overgrowth
Bone marrow and solid tissue neoplasms consist of cell types that proliferate spontaneously in culture, although often at a low rate. Lymph nodes are composed of cells that have a low intrinsic proliferative rate but that can be induced to divide much more rapidly by the addition of mitogens. Phytohemagglutinin (PHA) stimulates proliferation of T-lymphocytes. Lipopolysaccharide (LPS), protein A, 12-O-tetradecanolyphorbol-13-acetate (TPA), Epstein-Barr virus, synthetic oligonucleotides, and pokeweed mitogen induce proliferation of B-lymphocytes, and are also required for successful culture of some leukemias and lymphomas of B-cell origin.
2. Culture maintenance. The length of in vitro culture depends on cell type. Since bone marrow cultures contain spontaneously proliferating cells, they can be harvested after only a 24- to 48-hour culture interval, if not directly after specimen collection. Peripheral blood cultures usually require a 72-hour culture interval. The growth rate of solid tissue specimens is difficult to predict; some solid tumors require culture periods of 2 weeks or longer.
3. Cell harvest. Colcemid, a synthetic analogue of colchicine (an alkaloid from the bulb of the Mediterranean plant Colchicum), prevents separation of sister chromatids and is used to block the proliferating cells in metaphase, thus allowing an accumulation of cells at metaphase stage. A hypotonic solution is then used to swell the cells so that, after fixation, the chromosomes are adequately spread for microscopic analysis.
Since cells in culture do not proceed through the cell cycle in synchrony, chemical synchronization of cell division is often required to obtain an acceptable mitotic index. A common chemical approach involves addition of excess thymidine, which stalls cells at the S-phase of the cell cycle by decreasing the amount of dCTP available for DNA synthesis. When the excess thymidine is removed (or the effect of excess thymidine is eliminated by the addition of deoxycytidine), normal DNA replication resumes, and the collective release of the cells from S-phase produces a transiently high mitotic index. Alternatively, 5-fluorodeoxyuridine (which inhibits the enzyme thymidylate synthetase) can be used to stall cells at the G1/S boundary; in this method, addition of thymidine releases the block.
4. Banding. The different techniques that can be used to stain metaphase chromosomes can be divided into two general categories: methods that produce specific alternating white and dark regions (bands) along the length of each chromosome and methods that stain only a defined region of specific chromosomes (Table 58.2). In general, the dark bands are gene-poor AT-rich regions, whereas the light bands are composed of gene-rich GC-rich regions. The quality of staining depends on several technical factors, including sufficient separation of the chromosomes in the metaphase spread to allow clear visualization. Although there are no internationally accepted standards for banding resolution, ideograms are used as reference points (e-Fig. 58.1).* Many countries, including the United States, Canada, UK, France, Japan, and Australia have established standards that specify the minimum requirements for the number and quality of cells that must be processed for chromosome analysis depending on sample type, although many cases require even more detailed analysis.
5. Microscopic analysis. The method used to stain the chromosomes dictates whether bright-field microscopy or fluorescence microscopy is used to visualize the chromosomes. Conventional photography has traditionally been used to produce high-resolution prints of the stained chromosomes, but
electronic imaging systems have now replaced conventional photographic processes.
TABLE 58.2 Major Chromosome Staining and Banding Techniques
Method
Staining pattern
Techniques that produce specific alternating bands along each chromosome
Giemsa banding (G-banding)
Dark bands are AT rich; light bands are CG rich
Quinacrine banding (Q-banding)
Bright regions are AT rich
Reverse banding (R-banding)
AT-rich regions stain lightly (have dull fluorescence), CG-rich regions staining darkly (have bright fluorescence)
4,6-Diamidino-2-phenylindole (DAPI) staining
DAPI binds AT-rich regions; produces a pattern similar to Q-banding
Techniques that stain selective chromosome regions
Constitutive heterochromatin banding (C-banding)
Stains heterochromatin (α-satellite DNA) around the centromeres; can also be used to demonstrate some inherited polymorphisms
Telomere banding (T-banding)
Technical variation of R-banding used to stain telomeres
Silver staining for nucleolar organizer regions (NOR staining)
Stains the NORs (which contain rRNA genes) on the satellite stalks of acrocentric chromosomes
Fluorescence in situ hybridization (FISH)
Staining pattern is dependent on the probe
6. The final step in cytogenetic analysis is the production of a karyotype, which consists of the chromosomal complement of the cell displayed in a standard sequence on the basis of size, centromere location, and banding pattern (e-Fig. 58.2).
D. Assay failure. Many of the common causes of failure to obtain a cytogenetic result (Table 58.3) can be avoided by careful selection of viable tissue with prompt specimen transport to the cytogenetics laboratory in the appropriate medium. Nonetheless, several causes of assay failure are inherent to in vitro culture and cannot be eliminated by even the most meticulous laboratory technique.
TABLE 58.3 Common Reasons for Failure of Traditional Cytogenetic Analysis
Culture failure
No viable cells present in the sample (necrotic tumor sample or improper specimen handling)
Inappropriate sample (peripheral blood without blasts is submitted instead of bone marrow)
Overgrowth by nonneoplastic cells
Overgrowth by a nonrepresentative clone of tumor cells
Microbial overgrowth
Post culture failure
Technical errors involving cell harvest, slide preparation, or staining
Misdiagnosis (an abnormality is overlooked, or an abnormality is incorrectly interpreted)
Cytogenetic analysis of solid tumors highlights a number of these intrinsic technical limitations. First, since benign solid tumors contain few mitotic cells, cultures are susceptible to overgrowth by nonneoplastic cells. Second, even high-grade malignant solid tumors often grow poorly in vitro, especially if grown without the appropriate culture medium and growth factor supplementation. Third, the number of neoplastic cells in a solid tumor sample can be difficult to determine on the basis of gross examination, and the material submitted to the cytogenetics laboratory may consist primarily of stromal cells and inflammatory cells. Fourth, the viability of the neoplastic cells is often uncertain; even tumor samples that are not grossly necrotic may contain predominantly nonviable tumor cells. Fifth, in vitro culture selects for subclones within the neoplastic population that have a growth advantage, and so the karyotype may not be representative of the entire neoplasm. Sixth, contamination is often unavoidable for samples collected in the frozen section area or gross room, or from specimens arising from anatomic sites normally colonized by bacteria, such as the oral cavity, gastrointestinal tract, and skin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cytogenetics
Cytogenetics
Shashikant Kulkarni
Hussam Al-Kateb
Catherine Cottrell
