TLR
Subcellular localization
Exogenous ligands
Origin of exogenous ligand
Endogenous ligands
TLR1/TLR2
Cell surface
Triacyl lipopeptides
Bacteria, viruses
Unknown
(Pam3CSK4)
TLR2/TLR6
Cell surface
Diacyl lipopeptides PGN
Bacteria, viruses
HSP-60, HSP-70, HSP-90
LTA
HMGB1
Zymosan
TLR3
Intracellular
dsRNA
Viruses
mRNA
Cell surface
Poly I:C
Stathmin
TLR4
Cell surface
LPS
Bacteria, viruses
HSP-22, HSP-60, HSP-70
Lipid A derivatives
HMGB1, fibronectin
Defensin 2, oxLDL
Tenascin C
TLR5
Cell surface
Flagellin
Bacteria
Unknown
TLR7
Intracellular
ssRNA Imidazoquinoline
Bacteria, viruses
Self-RNA
Cell surface
Loxoribine
MicroRNA
Bropirimine
TLR8
Intracellular
ssRNA
Viruses
Self-RNA
Imidazoquinoline
MicroRNA
TLR9
Intracellular
Unmethylated CpG DNA
Bacteria, viruses, protozoa
Self-DNA
CpG ODNs
HMGB1
TLR10
Intracellular
Unknown
Unknown
Unknown
TLR11
Cell surface
Uropathogenic bacteria
Bacteria
Unknown
Profilin-like molecules
Protozoa
1.2 Ligands
Each type of TLR detects distinct molecular patterns specific to pathogenic threats to the organism such as viruses, bacteria, mycobacteria, fungi, and parasites. For example, TLR4 recognizes LPS (Poltorak et al. 1998; Shimazu et al. 1999), a component of Gram-negative bacterial cell membranes; TLR5 detects flagellin, a protein in bacterial flagella (Hayashi et al. 2001); and TLR11 senses profilin-like protein (Yarovinsky et al. 2005). TLR2 heterodimers bind to specific lipopeptides, another component of bacterial cell membranes. The ligand specificity of TLR2 is modulated by its heterodimeric partner. The TLR1–TLR2 complex binds to triacyl lipopeptides with only a weak affinity for diacyl lipopeptides, while the TLR2–TLR6 complex interacts with diacyl lipopeptides but not to triacyl lipopeptides (Takeuchi et al. 2001, 2002; Alexopoulou et al. 2002; Yamamoto et al. 2002; Jin et al. 2007; Kang et al. 2009). TLR3 senses double-stranded RNAs and TLR7/TLR8 single-stranded RNAs, present in retroviruses and viruses, respectively (Alexopoulou et al. 2001; Diebold et al. 2004; Heil et al. 2004; Town et al. 2006). TLR9 senses unmethylated CpG DNA (Hemmi et al. 2000; Krieg 2002). Importantly, TLRs also recognize endogenous markers of cell necrosis and tissue injury, termed DAMPs. Activation of TLRs by DAMPs induces sterile inflammatory responses (Okamura et al. 2001; Biragyn et al. 2002; Vabulas et al. 2002; Kariko et al. 2004; Jiang et al. 2005; Tian et al. 2007; Imai et al. 2008; Midwood et al. 2009; West et al. 2010). Thus, TLRs can recognize both pathogen invasion through their recognition of PAMPs and tissue injury through their recognition of endogenous DAMPs (summarized in Table 1).
1.3 Signaling Pathways
Activation of TLR signaling leads to a tightly regulated intracellular signaling pathway that initiates the production and secretion of proinflammatory mediators. The intracellular signaling domains of TLRs have a high sequence similarity with the interleukin-1 receptor and are termed Toll/IL-1R (TIR) homology domains. Signaling adaptor proteins MyD88, TRIF, and TRAM also contain TIR domains and interact with the TIR domains of the TLR receptors through heterotypic TIR–TIR interactions (Watters et al. 2007; Kenny and O’Neill 2008). TRIF and TRAM are also referred to as TICAM-1 and TICAM-2, respectively. All TLRs, with the exception of TLR3, signal through MyD88. TLR3 signals through a TRIF-dependent pathway discussed later. TLR4 is also capable of signaling in a MyD88-independent manner by recruiting TRAM (Yamamoto et al. 2003). TLR1/TLR2, TLR4, and TLR6 recruit the additional adapter protein TIRAP (Akira and Takeda 2004). Upon activation of the MyD88-dependent pathway, MyD88 recruits the IL-1R-associated kinases (IRAKs), which interact with tumor necrosis factor receptor-associated factor 6 (TRAF6), leading to the phosphorylation and degradation of the inhibitor of nuclear factor-κB (NF-kB) IκB. Degradation of IκB releases NF-κB which translocates to promote transcription of proinflammatory genes. The MyD88-dependent pathway also activates the mitogen-activated protein kinase (MAPK) signaling pathways, such as p38, and c-Jun N-terminal kinase (JNK), leading to the activation of AP-1 and interferon regulatory factors (IRFs) (see Fig. 1).
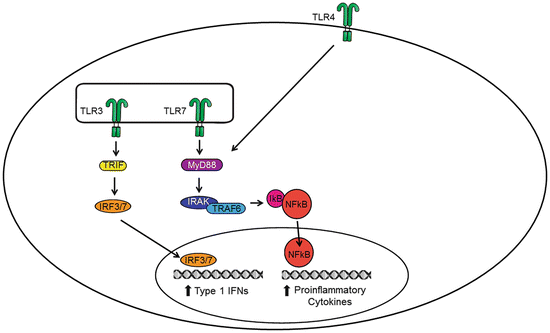
Fig. 1
Conventional signaling of TLRs. Activation of extracellular TLRs (e.g., TLR4) and intracellular TLRs (e.g., TLR3 and TLR7) initiates canonical TRIF- and MYD88-dependent signaling pathways resulting in the transcription of proinflammatory genes. Specifically, the activation of the transcriptional factors IRF3/IRF7 and NF-κB induces the production of inflammatory mediators such as cytokines (e.g., IL-1β, interferons), chemokines (e.g., CCL2), PGE2 via the enzyme cyclooxygenase 2 (COX-2), and NO via the enzyme inducible nitric oxide synthase (iNOS). These inflammatory mediators act in concert to initiate the host’s innate immune response
TLR3 signals through a TRIF-dependent pathway which activates two signaling pathways. First, TRIF recruits TRAF3 to activate IRF3 and IRF7 and initiate the production of type I interferons (e.g., IFN-α/IFN-β), which are the first line of defense produced by the innate immune system. Second, TRIF interacts with receptor-interacting protein 1 (RIP-1) and TRAF6 to activate NF-κB and/or MAPK pathways (Fig. 1). This is involved in the late-phase induction of proinflammatory genes.
The signaling molecules that comprise each TLR signaling pathway are also subject to a high degree of regulation through physical interactions, conformational changes, phosphorylation, ubiquitination, and proteasome-mediated degradation (Carpenter and O’Neill 2009). Some miRNAs are capable of regulating TLR signaling through targeting the 3′ untranslated regions of mRNAs encoding components of the TLR signaling system (O’Neill et al. 2011). Activation of TLR signaling produces a wide array of proinflammatory mediators, such as cytokines, chemokines, and reactive oxygen/nitrogen intermediates including nitric oxide (Takeda and Akira 2004). Upregulated activity of TLR signaling may promote chronic itch (Liu and Ji 2013).
2 Introduction to Itch
Itch, or pruritus, is defined as an unpleasant sensation that elicits the desire or reflex to scratch. While acute itch is an adaptive mechanism to warn an organism of potential chemical or parasitic danger (Ikoma et al. 2006), chronic itch is a common clinical problem associated with skin diseases (Reich and Szepietowski 2007; Bieber 2008), liver and kidney diseases (Cassano et al. 2010; Kremer et al. 2010), and metabolism disorders (Yamaoka et al. 2010). Additionally, itching itself leads to scratching which can cause inflammation, skin degradation, and secondary infection.
While some itch-sensing circuitry may overlap with existing nociceptive circuits, there are features that have recently been shown that are specific to itch. Itch sensation arises from MrgprA3+ fibers, derived from a specific subset of primary sensory neurons that reside in the skin, but not in deeper tissues, muscles, organs, or bones where itch sensation does not occur (Han et al. 2013). Their cell bodies are located in the dorsal root ganglia (DRG) or trigeminal ganglia, while their peripheral terminals, free nerve endings, reach to the stratum granulosum of the epidermis, and their central projections terminate in the superficial horn of the spinal cord. Itch-specific neurons terminate in lamina I–II and release glutamate as well as gastrin-releasing peptide (GRP), a neuropeptide known to elicit itch sensation via activation of GRP receptors (GRPRs) on superficial dorsal horn neurons (Sun and Chen 2007; Liu et al. 2009; Sun et al. 2009). A major source of GRP in the spinal cord may arise from interneurons (Alemi et al. 2013; Mishra and Hoon 2013). In addition, the neuropeptide natriuretic polypeptide b (Nppb) and substance P are also implicated in spinal cord itch transmission (Akiyama and Carstens 2013; Mishra and Hoon 2013). In the dorsal horn, itch signals can be processed and modulated before ascending to the brain where activation of specific brain regions then results in the perception of itch. Notably, in acute conditions, pain can suppress itch sensation via spinal cord inhibitory neurons (Liu et al. 2010b; Ross et al. 2010; Liu and Ji 2013).
The most characterized mediator of itch is histamine. The released histamine from local mast cells binds to H1/H4 receptors on nerve terminals (Shim and Oh 2008) which is followed by activation of PLCbeta3 and transient receptor potential vanilloid subtype 1 (TRPV1) (Han et al. 2006; Imamachi et al. 2009). TRPV1-positive C-fibers are required for both histamine-dependent and histamine-independent itch (Imamachi et al. 2009). Histamine-independent itch can be induced by the activation of transient receptor potential cation channel, subfamily A, member 1 (TRPA1), chloroquine (an antimalarial drug and MrgprA3 agonist), BAM8–22 (an endogenous MrgprC11 agonist), and oxidative stress (Liu et al. 2009; Wilson et al. 2011; Liu and Ji 2012), respectively.
3 Control of Itch by Peripheral TLRs
The skin is the body’s largest organ and first line of defense against microbial and parasitic invaders. The skin expresses every type of known TLR receptor; however, each cell type has a unique expression pattern and distinct contribution to the skin’s immune response (Ermertcan et al. 2011). In particular, two TLR-expressing cell type keratinocytes and mast cells have been implicated in chronic itch (Ikoma et al. 2006) (see Fig. 2).
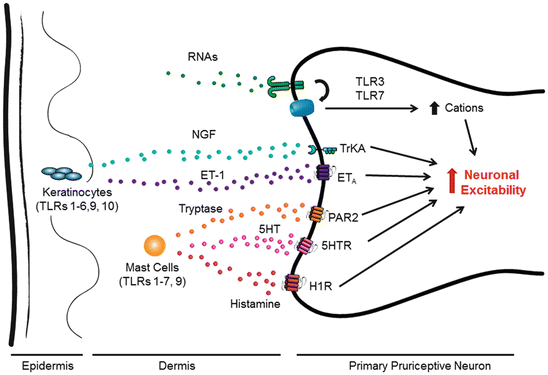
Fig. 2
Involvement of peripherally expressed TLRs in itch sensation via different mechanisms. Cells residing in the skin such as keratinocytes and mast cells express TLRs. Their activation by exogenous ligands (PAMP) and endogenous ligands (DAMP) results in the release of multiple pruritogens including NGF, ET-1, 5HT, tryptase, and histamine, all of which can activate G protein coupled receptors (GPCRs) or TrkA receptor (in the case of NGF) on pruriceptive neuronal terminals to increase neuronal excitation and trigger itch sensation. TLRs such as TLR3 and TLR7 are also expressed by pruriceptor terminals, which can be activated by DAMP such as double-strand and single-strand RNAs. Activation of TLR3 and TLR7 elicits rapid inward currents and action potentials, due to the direct coupling with ion channels in pruriceptors, leading to increased neuronal excitation and itch sensation. A second late-phase response to the activation of TLRs may also involve the increased transcription of proinflammatory mediators which can maintain neuronal hyperexcitability in chronic itch conditions
Epidermal keratinocytes express TLRs 1–6, 9, and 10, which are upregulated in pruritic skin diseases, such as psoriasis and atopic dermatitis (Baker et al. 2003; Ermertcan et al. 2011). Mast cells, predominantly in the dermis, express TLRs 1–7 and 9 and play a key role in IgE-mediated allergic inflammation. Activation of these TLRs results in the synthesis and release of cytokines and chemokines to recruit immune cells from the circulation and mount an adaptive immune response. In particular, keratinocytes and mast cells are the major sources of nerve growth factor (NGF) (Ikoma et al. 2006). Intradermal administration of NGF enhances itch sensation in humans (Rukwied et al. 2013), and NGF is upregulated in dry skin models of itch in mice (Tominaga et al. 2007). Of interest, this increase is suppressed in Tlr3 knockout animals (Liu et al. 2012). Therefore, TLR3-mediated release of NGF from skin cells may contribute to peripheral sensitization in chronic itch conditions.
While TLRs have mainly been studied on immune cells, we have recently demonstrated the expression of TLR3 and TLR7 on a subset of primary sensory neurons that are responsible for itch. Single-cell Real-time PCR (RT-PCR) and immunohistochemistry demonstrated that TLR7 is expressed in small TRPV1-expressing DRG neurons (Liu et al. 2010a, 2012) and is completely colocalized with MrgprA3+. TLR3 partially colocalizes with TLR7 and is also expressed in TRPV1-positive DRG neurons as shown by in situ hybridization, immunocytochemistry, single-cell RT-PCR, and electrophysiology (Liu et al. 2012).
TLR7 responds to synthetic ligands such as imidazoquinoline derivatives [e.g., imiquimod and resiquimod (R848)] and guanine analogs (e.g., loxoribine) (Hemmi et al. 2002). Intradermal injection of these synthetic compounds produces scratching behavior in wild-type mice, which is reduced in Tlr7 −/− mice, suggesting that these responses are, at least partially, TLR7 dependent (Liu et al. 2010a). Tlr7 −/− mice also show a significant reduction of scratching behavior in response to non-histaminergic pruritogens such as chloroquine, endothelin-1 (ET-1), and SLIGRL-NH2, an agonist of protease-activated receptor 2 (PAR2) (Liu et al. 2010a). Interestingly, Tlr7 −/− mice exhibited normal thermal and mechanical pain sensitivity. These findings show that TLR7 may serve an important role as a non-histaminergic itch receptor.
Activation of TLR7 in immune cells initiates signaling cascades leading to a variety of transcriptional changes to promote inflammation. However, in small DRG neurons, application of TLR7 agonists elicits rapid inward currents and action potentials, which are abolished in Tlr7 −/− knockout mice (Liu et al. 2010a). This immediate increase in neuronal excitability suggests that TLR7 may be coupled to ion channel activation in primary sensory neurons. Our recent study shows that TLR7 is functionally coupled to TRPA1 but not TRPV1 (Park et al. 2014).
TLR3 responds to dsRNA as well as its synthetic analog, polyinosinic–polycytidylic acid (poly I:C), in which one strand of RNA is replaced by a polymer of inosinic acid. Activation of TLR3 by its ligands poly I:C or purified total RNAs also elicits rapid inward currents and the generation of action potentials from DRG neurons in wild-type but not Tlr3 −/− mice (Liu et al. 2012). Furthermore, intradermal application of poly I:C produced dose-dependent scratching behavior in wild-type mice which was abolished in Tlr3 −/− mice (Liu et al. 2012). Similar to TLR7, TLR3 also seems to serve as an itch receptor/co-receptor on pruriceptive neurons. In contrast to Tlr7 −/− mice, which showed a partial reduction in histamine-independent itch, Tlr3 −/− mice displayed significant reductions in both histamine-dependent and histamine-independent itch (Liu et al. 2012). Knockdown of TLR3 by intrathecal injection of Tlr3 antisense oligonucleotides significantly reduced TLR3 expression in the DRG and reduced both histamine-dependent and histamine-independent itch in wild-type mice (Liu et al. 2012). This corroborates the results found using Tlr3 knockout animals.
TLR3 and TLR7 expression by primary sensory neurons seems to serve as itch receptors to detect foreign pathogens and endogenous ligands (e.g., ds- and ssRNA, respectively), leading to a rapid defensive response: scratching. As neuronal excitation occurs within minutes of agonist application, the neuronal signaling pathway must have a distinct component from the traditional TLR signaling pathway in immune cells; however, the details remain to be determined. We postulate that TLR3 and TLR7 are expressed on the cell surface and are coupled to unidentified ion channels capable of inducing inward currents and action potentials (see Fig. 2). Of note, it was also demonstrated that the activation of TLRs, including TLR3, TLR7, and TLR9, in DRG neurons by their respective ligands may indirectly influence the excitability of DRG neurons by inducing the expression of proinflammatory mediators such as prostaglandin E2 (PGE2), calcitonin gene-related peptide (CGRP), and interleukin-1beta (IL-1β) (Qi et al. 2011) (see Table 2). Thus, TLRs expressed by DRG neurons may regulate neuronal excitability by both transcriptional and non-transcriptional mechanisms.
Table 2




Distribution of TLRs in the spinal cord and DRG
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
