Congenital Diaphragmatic Hernia
Charles J.H. Stolar
Ari R. Reichstein
For several reasons, there are few conditions that are as enigmatic for pediatric surgeons as congenital diaphragmatic hernia (CDH). First, despite intense basic and clinical research as well as innovative new technologies, the morbidity and mortality for this condition remain significant. Second, the most significant effect on survival has actually been a strategy of less aggressive ventilation and invasive intervention. Third, the ability to predict outcome probability for any given infant with this anomaly has remained elusive. Finally, it must be underscored that the operative repair of CDH is one of the least important factors affecting patient survival. This chapter focuses primarily on the most common posterolateral defect of Bochdalek. The less common retrosternal hernia of Morgagni is also described.
History
The earliest English-language description of the anatomy and pathophysiology associated with CDH in a newborn was presented by McAuley in 1754. The actual defect was named following the description of this same abnormality by Bochdalek in 1848. Successful repair of CDH remained rare until 1940, when Ladd and Gross
reported 9 of 16 patients surviving surgery, with the youngest being 40 hours old. It was not until 1946 that Gross reported the first infant who was less than 24 hours old and who survived operative repair. Immediate neonatal repair of the defect followed by postoperative resuscitative therapy became the standard treatment for CDH. It was felt that the lung compression and mediastinal shift caused by the herniated viscera was a significant contributor toward mortality. It was not until the mid-1980s that there was a paradigm shift in understanding the greater importance of the underlying pulmonary hypoplasia and degree of hypertension rather than the herniated viscera. This understanding, coupled with the realization that early operative intervention in these tenuous patients was probably more harmful, has directed the more accepted approach of delayed surgery following a period of stabilization.
reported 9 of 16 patients surviving surgery, with the youngest being 40 hours old. It was not until 1946 that Gross reported the first infant who was less than 24 hours old and who survived operative repair. Immediate neonatal repair of the defect followed by postoperative resuscitative therapy became the standard treatment for CDH. It was felt that the lung compression and mediastinal shift caused by the herniated viscera was a significant contributor toward mortality. It was not until the mid-1980s that there was a paradigm shift in understanding the greater importance of the underlying pulmonary hypoplasia and degree of hypertension rather than the herniated viscera. This understanding, coupled with the realization that early operative intervention in these tenuous patients was probably more harmful, has directed the more accepted approach of delayed surgery following a period of stabilization.
Embryology of the Diaphragm and Lung
The classic embryologic basis for CDH remains focused on the formation of the diaphragm, which takes place during the 4th through the 8th weeks of gestation. The complete diaphragm is derived from several components: (a) the septum transversum that forms the central tendon; (b) bilateral pleuroperitoneal membranes or folds that are reinforced by striated muscle components; and (c) the mesentery of the esophagus, which forms crural and dorsal structures. A long-standing hypothesis has been that incomplete or absent fusion of the pleuroperitoneal folds separating the chest from the abdomen is responsible for the posterolateral defect.
Adequate intrathoracic space is a prerequisite for normal lung growth, and any intrathoracic or extrathoracic process that impairs this space results in pulmonary parenchymal compression and structurally immature lungs. Closure of the pleuroperitoneal canals with the formation of a pleuroperitoneal membrane occurs during the 8th week of gestation. This is facilitated by pleuroperitoneal folds that extend from the lateral body wall and grow medially and ventrally until they fuse with the septum transversum and dorsal mesentery of the esophagus during gestational week 6. Anatomically, the right side closes before the left, and additional muscularization of the diaphragm appears to develop from the innermost muscle layer of the thoracic cavity.
In addition to embryologic development of the diaphragm, it is important to understand related fetal lung development. This is divided into five stages: embryonic, pseu-doglandular, canalicular, saccular, and alveolar. Embryonic lung development begins during the 3rd week of gestation as a derivative of the foregut and is marked by the formation of a diverticulum from the caudal end of the laryngotracheal groove. The trachea and the two primary lung buds develop from this diverticulum by the 4th week. By 6 weeks, these lung buds have further developed into defined lobar structures. The pseudoglandular phase of lung development is the period for bronchial airway development and takes place during the 7th to 16th weeks of gestation. From the 16th through the 24th week of gestation, the canalicular phase of growth occurs, and during this period air space development is noted as crude alveolar air sacs are identified. It is during this phase that type I pneumocytes begin to differentiate and precursors of type II pneumocytes, which are ultimately responsible for surfactant production, begin to appear. There is continued maturation of the alveolar air spaces during the saccular phase of development, extending beyond 24 weeks of gestation. After 24 weeks, there is continued remodeling of the air space dimensions and maturation of surfactant synthesis. True adult-like alveoli begin to appear shortly following birth, and continued lung development with alveolar maturation and multiplication continues until about 8 years of age.
While the traditional embryologic basis for CDH holds that visceral compression of the developing lung is the inciting event, emerging evidence supports the theory that pulmonary hypoplasia may precede diaphragmatic agenesis. It remains unclear whether impaired pulmonary development causes the diaphragmatic defect or whether both conditions are attributable to a common proximate cause. A “dual hit” hypothesis has been proposed, with the first hit consisting of primary pulmonary hypoplasia, and a second insult resulting from mechanical visceral compression. This model accounts for the finding that pulmonary hypoplasia is bilateral but frequently more severe ipsilateral to the diaphragmatic defect.
Genetic Factors and Associated Anomalies
The cause of CDH is currently unknown but is likely to be multifactorial, encompassing both environmental and genetic factors. Traditionally, CDH has been classified as isolated (nonsyndromic) or nonisolated (syndromic). Approximately 60% of cases are categorized as isolated defects. This classification can be misleading, because even isolated CDH frequently presents with associated anomalies related to the hernia’s developmental field defect. Such anomalies include patent ductus arteriosus, an unfixed midgut, and esophageal ectasia. Approximately 40% of cases are associated with non–hernia-related malformations and are categorized as nonisolated CDH. The most frequently identified syndrome in these cases is Fryns syndrome, although several other monogenic syndromes featuring CDH have been described. Regardless of whether the defect is categorized as isolated or nonisolated, associated congenital heart anomalies (such as hypoplastic heart or transposition of the great arteries) and/or brain malformations suggest greater severity of disease and portend a worse prognosis.
Congenital Posterolateral Diaphragmatic Hernia of Bochdalek
Incidence
The reported incidence of CDH is estimated to be between 1 in 2,000 to 5,000 births. The incidence in stillborns is less well documented and often associated with other major anomalies. Defects are much more common on the left side, with approximately 80% being on the left and the remainder on the right. Bilateral CDH defects are quite rare. Although this is typically considered to be a sporadic developmental anomaly, a number of familial and twin pregnancy cases have been reported.
The classic left-sided CDH features a 2- to 4-cm posterolateral defect in the diaphragm through which the abdominal viscera are translocated into the hemithorax (Fig. 1). The defect is usually seen as a completely open space between the chest and abdomen, although a sac is identified in a small percentage (<10%) of patients. The herniated abdominal contents in the left chest often include the left lobe of the liver, the spleen, and variable segments of the gastrointestinal tract. The stomach is frequently up in the chest, resulting in some degree of obstruction at the gastroesophageal junction. This obstruction results in polyhydramnios, with dilation and ectasia of the esophagus. In a right-sided defect, the large right lobe of the liver is capable of occupying much of the hemithorax in addition to other abdominal viscera.
Fetal animal studies have demonstrated that long-term compression of the developing
lungs by herniation of abdominal contents or paralysis of the diaphragm results in pulmonary vascular maldevelopment and lung hypoplasia. Whether pulmonary hypoplasia or hypertension predominates determines the clinical presentation and long-term outcome of this process. It is important to understand that although the ipsilateral lung is more severely affected than the contralateral lung, both are histologically abnormal. Because the process of CDH herniation probably occurs at the time of bronchial subdivision (pseudoglandular phase), it is at this stage that the lung development becomes perturbed. Although all major bronchial buds are present in the most affected lung, the number of bronchial branches is greatly reduced. The number of alveoli per bronchiole is actually increased, perhaps as a compensatory response.
lungs by herniation of abdominal contents or paralysis of the diaphragm results in pulmonary vascular maldevelopment and lung hypoplasia. Whether pulmonary hypoplasia or hypertension predominates determines the clinical presentation and long-term outcome of this process. It is important to understand that although the ipsilateral lung is more severely affected than the contralateral lung, both are histologically abnormal. Because the process of CDH herniation probably occurs at the time of bronchial subdivision (pseudoglandular phase), it is at this stage that the lung development becomes perturbed. Although all major bronchial buds are present in the most affected lung, the number of bronchial branches is greatly reduced. The number of alveoli per bronchiole is actually increased, perhaps as a compensatory response.
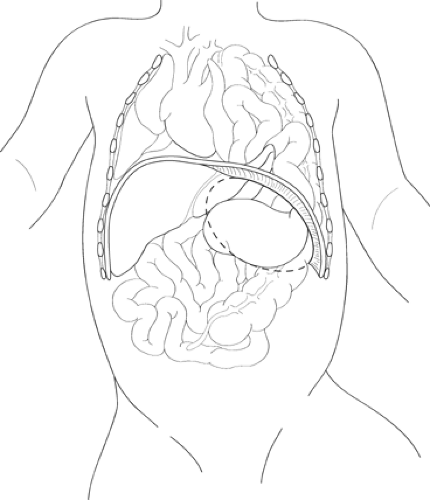 Fig. 1. Left-sided congenital diaphragmatic hernia involving the stomach, left lobe of the liver, small bowel, and colon translocated into the left hemithorax. The mediastinum is displaced to the right. The defect in the diaphragm is indicated by the dashed line. |
Bearing in mind that the pulmonary artery grows “hand in glove” with the developing airway, the pulmonary vascular bed in infants with CDH is distinctly abnormal. There is a reduction in the total number of arterial branches in both the ipsilateral and contralateral sides. In addition, the small pre- and intra-acinar arterioles show inappropriate and significant medial muscular hyperplasia. The consequence of this is an abnormal increased susceptibility to the development of pulmonary hypertension. Moreover, the cross-sectional surface area of the pulmonary arteries is also reduced, yielding a fixed lesion even after remodeling. In addition to compression of the ipsilateral lung, there is usually mediastinal compression with a shift of the heart and pericardial structures to the right, thus also compressing the opposite lung. As previously mentioned, virtually all patients have an unfixed midgut in addition to esophageal dilation and ectasia. This is not to be confused with classic malrotation and midgut volvulus.
Pathophysiology
The two primary factors in the pathophysiology of CDH are pulmonary hypoplasia and pulmonary hypertension resulting in inadequate gas exchange and diminished pulmonary blood flow. Severe pulmonary hypoplasia may be overwhelming and lethal. On the other hand, pulmonary hypertension is a potentially reversible component of this anomaly. As described previously, the walls of the arterials are extremely muscular and highly sensitive to factors that control pulmonary blood flow. The most significant factors to induce pulmonary artery vasoconstriction include acidosis, hypoxia, and hypercarbia. A transient fall in Pao2 or a brief episode of respiratory acidosis with elevated PaCO2 will trigger pulmonary vasoconstriction that causes an even more hypoxic environment. This generates an even greater pulmonary vasospastic response. Major interventions are therefore designed to prevent hypoxemia, acidosis, and hypercarbia as well as to consider pharmacologic measures to induce selective pulmonary vasodilation. Again, despite postnatal vascular remodeling, the reduced cross-sectional area of the pulmonary artery may prove to be a rate-limiting factor for clinical improvement.
Historically, aggressive ventilation strategies have been employed to normalize both Pao2 and PaCO2 levels. It is now realized that this is associated with significant barotrauma and is not necessary. More recent strategies have focused on adequate levels of preductal oxygenation—not completely normalized—to maintain a balance between provision of sufficient oxygen delivery and minimization of barotrauma. This newer strategy is termed permissive hypercapnia/spontaneous ventilation. The goals for support with these modalities include maintaining preductal o2 saturations greater than 90% and PaCO2 levels less than 60 mm Hg.
With increasing frequency, CDH is diagnosed in the antenatal period by ultrasonography. This anomaly is identified either during routine screening or during an evaluation of polyhydramnios. Ultrasonography demonstrates mediastinal shift away from the side of the CDH, with loops of intestine, stomach, or liver within the thoracic cavity. The polyhydramnios is due to the obstructed distal esophagus secondary to the acute esophagogastric angle due to the stomach being translocated into the chest. Often, prenatal magnetic resonance imaging can confirm the diagnosis and reveal additional anomalies.
CDH can be diagnosed in a range of postnatal presentations. For example, the child may present with severe respiratory distress from the outset, with grunting, intercostal and suprasternal retractions, and marked cyanosis, requiring urgent treatment. Alternatively, the child is born with a normal respiratory pattern, minimal cyanosis, and few detectable anomalies. After 6 to 8 hours, however, the child develops progressive respiratory distress, with retractions, grunting, and increased work of breathing. An unusual presentation of CDH later in life is uncommon, but is associated with the best survival. These children have no demonstrable respiratory distress and typically present later in life with bowel obstruction
due to the enteric contents in the chest, or an incidental finding seen on a routine chest radiograph. Other physical findings of CDH include a scaphoid abdomen with a barrel-shaped, asymmetric chest. Breath sounds are diminished on the ipsilateral side. Occasionally, bowel sounds may be heard in the affected hemithorax. However, because the patient is frequently mechanically ventilated, it is usually not possible to hear bowel sounds in the chest.
due to the enteric contents in the chest, or an incidental finding seen on a routine chest radiograph. Other physical findings of CDH include a scaphoid abdomen with a barrel-shaped, asymmetric chest. Breath sounds are diminished on the ipsilateral side. Occasionally, bowel sounds may be heard in the affected hemithorax. However, because the patient is frequently mechanically ventilated, it is usually not possible to hear bowel sounds in the chest.
Aside from prenatal ultrasound, the diagnosis of CDH is usually easily made on a plain anterior–posterior chest radiograph with intestinal contents identified within the affected hemithorax. The tip of the nasogastric tube may well be identified within the ipsilateral hemithorax along with the stomach. Occasionally, cystic lung lesions (congenital cystic adenomatoid malformation or multiple lung cysts) may be confused with air-containing loops of intestine. Another entity to consider is eventration of an intact diaphragm. In an otherwise stable patient, a contrast upper gastrointestinal series may help to clarify the diagnosis of CDH.
If the diagnosis of CDH has been established prenatally, arrangements should be made to deliver the child at the highest-level neonatal facility available. Availability of extracorporeal membrane oxygenation (ECMO) is essential. Infants in whom CDH is postnatally diagnosed should be immediately referred to an appropriate tertiary-care children’s hospital following delivery. A spontaneous vaginal delivery is preferred unless other obstetrical issues supervene. At present, fetal intervention for in utero correction of CDH is not advocated.
After birth of the infant and confirmation of the diagnosis, all efforts should be made to stabilize the cardiorespiratory system while minimizing iatrogenic injury. It should be underscored that CDH is a physiologic but not a surgical emergency. Establishment of an appropriate airway consists of nasotracheal intubation, administration of supplemental oxygen, and nasogastric tube insertion. Effective nasogastric decompression is crucial to prevent further distention of the viscera within the chest and additional cardiovascular compromise. Ventilation by mask alone is contraindicated, as this will result in gastric distention and further mediastinal and lung compression.
Intravenous and arterial access should be readily obtained. This is usually done through the umbilical vein, umbilical or peripheral preductal artery, and peripheral venous lines. Metabolic acid-based disturbances are usually related to hypoperfusion and should be corrected by appropriate fluids and administration of bicarbonate. A urethral catheter should be placed for accurate measurement of urine output.
The type of mechanical ventilator needed for the infant with CDH is a matter of personal and institutional preference. Simple continuous flow pressure-cycled ventilator therapy with modest peak airway pressures (18 to 22 cm H2O) may be used, with a goal of obtaining a preductal Pao2 saturation above 90%. In addition, the PaCO2 should be less than 60 mm Hg. Inability to achieve sustained preductal saturations above 90% with aggressive ventilator strategies is usually associated with significant pulmonary hypoplasia and a lethal course. A broad spectrum of pharmacologic antihypertensive agents have been employed to reduce pulmonary vascular resistance. Unfortunately, most have the detrimental side effect of systemic hypotension, resulting in reduced cardiac output and worsening of systemic oxygenation and oxygen delivery. Nitric oxide was originally felt to be an important strategy to cause selective pulmonary vasodilation; however, the effect of nitric oxide on infants with CDH has been modest at best.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


