and FJE Vajda2
(1)
Clinical Neurology and Neuropharmacology, University of Queensland, and Honorary Consultant Neurologist, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
(2)
Department of Medicine and Neurology Director of the Australian Epilepsy and Pregnancy Register, University of Melbourne and Royal Melbourne Hospital, Melbourne, Australia
Abstract
Data are available in the literature for the clinical pharmacologies of three newer antiepileptic drugs which have achieved a moderate amount of use in pregnant women, viz. lamotrigine, topiramate and levetiracetam. There is more information available for the first of these drugs than for the other two. The clearance value for lamotrigine, which is almost fully biotransformed to glucuronides, is considerably increased during pregnancy, with the clearances of topiramate (eliminated by a mix of metabolism and renal excretion unchanged) and levetiracetam (eliminated mainly by renal excretion without prior metabolism) being less increased. Relatively little information is available concerning the dispositions of the three drugs in the neonate, though there are published data for maternal plasma to breast milk concentration ratios.
At the time of writing, several of the antiepileptic drugs that have been introduced into therapeutic practice in the past quarter of a century seem to have become used increasingly by pregnant women, with apparently satisfactory outcomes. The clinical pharmacologies of the three most commonly encountered of these agents are discussed below.
Lamotrigine
Lamotrigine was developed as an antiepileptic agent after it was noticed that use of high doses of the older antiepileptic drugs phenobarbitone and phenytoin was associated with reduced circulating folate concentrations. This knowledge suggested that lamotrigine , with a molecular structure resembling part of the folate molecule, might have antiepileptic effects by interfering with the actions of folate. This has proved not to be lamotrigine’s mechanism of antiepileptic action. Nevertheless, the drug has been found effective in treating many of the commonly occurring varieties of epilepsy. Lamotrigine has a reasonably trouble-free adverse effect profile and, at least in more affluent countries, has come into rather extensive use both in treating epilepsy and in managing certain psychiatric disorders.
Chemistry
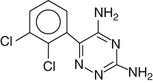
Lamotrigine [3,5-diamino-6-(2,3-dichlorophenyl)-1,2,4-triazine] is a white solid with a relatively poor aqueous solubility, a molecular weight of 256.09 and a pK value of 5.7. It is marketed in 5, 25, 50, 100 and 200 mg solid dosage forms.
Pharmacodynamics
As mentioned above, lamotrigine does not act via folate -related mechanisms. Like phenytoin and carbamazepine , its antiepileptic effect is achieved by blocking voltage-, use- and frequency-dependent Na+ ion channels in cell membranes. In doing this, lamotrigine in effect discriminates against the passage of rapid trains of axon impulses while interfering less with axon impulse transmission at more physiological frequencies. This mode of action reduces the release of neurotransmitters at axon terminals, particularly that of the excitatory molecule glutamate . Thus the drug’s mechanism of action is hardly different from those of the other Na+ channel-blocking antiepileptic agents phenytoin and carbamazepine. However, lamotrigine also has some inhibitory effects on voltage-activated Ca2+ currents and on K+ conductances . The drug’s action on Na+ channels may explain its effectiveness in controlling focal epilepsies and the generalised convulsive seizures of genetic generalised epilepsy . However, the known biochemical mechanisms of action of the drug do not seem capable of explaining why it can also be effective against absence seizures and against some forms of myoclonic seizure (though there are reports that it can make the latter worse).
Pharmacokinetics
The pharmacokinetic parameters that are quoted below apply for lamotrigine when used as the sole antiepileptic agent. In clinical practice the drug is not infrequently prescribed in combination with other antiepileptic drugs. The various parameters may then have different values, as will be mentioned when the pharmacokinetic interactions that involve the drug are considered.
Absorption
Lamotrigine is nearly always administered by mouth and seems to have a virtually complete oral bioavailability. Peak plasma levels of the drug occur 1–3 h after intake.
Distribution
The drug has an apparent volume of distribution of around 1.2 or 1.3 L per kg, consistent with its being distributed throughout body water and also achieving some higher local concentration somewhere in body tissues. Lamotrigine ’s concentrations in the brain are calculated to be about 2.8 times of those in serum. Approximately 55 % of the lamotrigine in plasma is bound to proteins, a figure which correlates with the drug’s concentration in cerebrospinal fluid (which averages 43 % of its plasma concentrations). Lamotrigine ’s concentration in saliva is also similar to its unbound concentration in plasma (Malone et al. 2006).
Elimination
Lamotrigine ’s elimination follows linear kinetics. Its half-life after an initial dose is approximately 33 h and in chronic use around 25 h. The drug’s first-dose clearance is 0.026 L per kg per hour, but the value is higher when the drug is in continued use. This is not a particularly high clearance value and does not suggest that the drug undergoes any extensive pre-systemic metabolism . The clearance is higher in children, some 0.038 L per kg per hour. There is no evidence that lamotrigine induces its own metabolism to any significant extent, and it does not induce the cytochrome P450 drug-metabolising enzyme system.
Excretion Unchanged
Very little unmetabolised lamotrigine is excreted in urine. In view of its complete apparent oral bioavailability, the drug therefore appears to be eliminated almost exclusively by metabolism.
Metabolism
A little more than 3/4 of a lamotrigine dose is excreted in urine as a 2-N-glucuronide , with a small amount as a 5-N-glucuronide and with something less than 1 % as a 2-N-methyl derivative. The glucuronidations are catalysed by UDP glucuronosyl transferase 1A4 (Chen et al. 2009). In Gilbert’s syndrome, with its decreased glucuronidation capacity, the clearance of lamotrigine is diminished. The glucuronide metabolites of lamotrigine appear to lack biological activity.
Clinical Pharmacokinetics
Lamotrigine concentrations can be monitored in plasma or serum or in saliva (Malone et al. 2006). There is a broad correlation between increasing plasma levels of the drug and increasing proportions of patients who achieve seizure control if they have types of epilepsy that normally respond to the drug. However the concentration range over which seizure control is achieved in the individual is quite wide. Early suggestions that the drug has a therapeutic range of 1–4 mg/L have been superseded by a proposed therapeutic set of values ranging between 1 and 15 mg/L. Patsalos et al. (2008) cited a range of 2.5–15 mg/L. Such a range is too wide to be helpful clinically in providing a preliminary indication that a therapeutically adequate dose of the drug has been prescribed. Nevertheless, the plasma lamotrigine concentration in the individual whose seizures are controlled provides a valuable indicator for guiding the future management of the seizure disorder in that particular patient, for instance, during pregnancy.
Interactions
The pharmacokinetic interactions in which lamotrigine is involved are mainly ones with agents that affect the glucuronidation of the drug, its main pathway of elimination .
Drugs which induce the capacity for glucuronidation , such as the older antiepileptic drugs phenobarbitone , phenytoin and carbamazepine and also rifampicin and lopinavir , may cause reduced lamotrigine plasma levels relative to the lamotrigine dose. As mentioned previously (Chap. 3), oestrogens may induce UDP-glucuronosyl transferases , and oestrogen -containing oral contraceptive use is associated with a tendency for plasma lamotrigine concentrations to be lower relative to the drug dose (Herzog et al. 2009; Wegner et al. 2009). This particularly is the case in the earlier parts of the menstrual cycle where the combined oral contraceptives provide oestrogen only. This same interaction with oestrogens contributes to the behaviour of plasma lamotrigine concentrations during the course of pregnancy.
Valproate inhibits the glucuronidation of lamotrigine and raises its plasma level relative to its dose. There have been reports that plasma lamotrigine levels may not fall during the course of pregnancy if valproate is also taken by the woman involved. Presumably, the effects of pregnancy and of the drug–drug interaction cancel each other. The magnitude of the interaction between valproate and lamotrigine in clinical practice is such that significantly lower than otherwise expected lamotrigine doses should be used if the drug is introduced into the treatment regimen of a patient already taking valproate.
Adverse Effects
Lamotrigine is not a particularly troublesome antiepileptic drug from the viewpoint of causing adverse effects.
Skin rashes may occur, nearly always quite early in the course of treatment. When the drug was first introduced into therapeutics, it was reported to cause a very fine, almost pinpoint, erythematous rash in the first few days of intake. If treatment with the drug was ceased and then resumed in lower dosage, this rash was reported not to recur. Sometimes rashes associated with the drug may be quite severe and even life-threatening, e.g. a Stevens–Johnson syndrome .
The drug does produce a degree of sedation which is dosage related and can be responsible for mental dulling, dizziness, blurred or double vision , drowsiness and ataxia of gait. Lamotrigine has a reputation for interfering less with sexual function than certain other antiepileptic agents.
Other adverse effects of the drug are uncommon.
Pregnancy
The Mother
While studying lamotrigine concentrations in breast milk and in the neonate, Rambeck et al. (1997) noticed that the drug’s concentration in maternal plasma relative to the drug dose fell during pregnancy and rose again by the third postpartum week, necessitating a reduction in the lamotrigine dose at that stage. In the same year, Tomson et al. (1997) followed the relationship between the lamotrigine dose and the circulating concentration of the drug throughout pregnancy and the postnatal period, thus obtaining data on the course of the steady-state apparent clearance of the drug. Compared with the situation that applied five months after giving birth, by late pregnancy the apparent clearance of lamotrigine had increased by a factor of 3.6 and at term was 5.8 times higher than postpartum. Tran (2002) then reported that lamotrigine’s clearance increased by 50 % during pregnancy. De Haan et al. (2004) later followed the relationship between circulating lamotrigine concentrations and drug dose during 12 pregnancies in women who took the drug in monotherapy. The ratio (i.e. the reciprocal of the steady-state apparent clearance ) fell by about 40 % during pregnancy and quickly returned to its pre-pregnancy value after the baby was delivered. The ratio reached its trough in the period between 20 and 30 weeks of pregnancy and began to rise again in the final 10 weeks. Pennell et al. (2004) followed plasma lamotrigine levels monthly throughout pregnancy in 14 women and found the drug’s apparent clearance increased (by more than 330 %) by the 32nd week of gestation and thereafter declined. Figure 5.1 shows the course of the apparent clearance of lamotrigine during and after pregnancy in one woman.
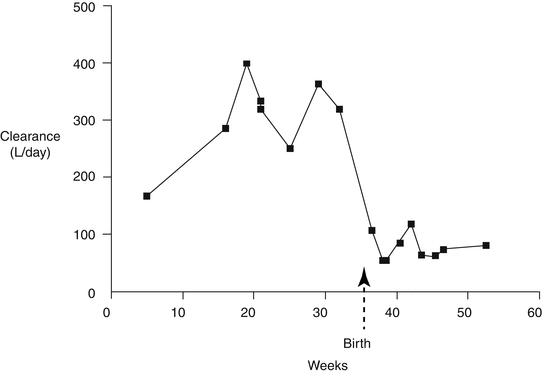
Fig. 5.1
The behaviour of the apparent clearance of lamotrigine during and after pregnancy in one woman
Thus, by about a decade after lamotrigine began to come into general clinical use, reasonably good evidence had become available that the clearance of the drug increased during human pregnancy.
Subsequent studies have confirmed the existence of this increased clearance when the drug is used in pregnancy in the absence of potentially interacting co-medication. Petrenaite et al. (2005), in 11 pregnancies, described a 65 % clearance increase in the second trimester with the increase remaining at 65.8 % in the third trimester. These authors commented on the degree of variation in the increase in the individual women. Franco et al. (2008) found a mean increased clearance of 164 % by late pregnancy, and Pennell et al. (2008) in 53 pregnancies found that both the total clearance and the clearance of unbound lamotrigine were increased in all trimesters of pregnancy, the peak increases being 94 and 89 % respectively in the third trimester. Fotopoulou et al. (2009) cited a 197 % increase in the first trimester, a 236 % increase by the second trimester and a 248 % increase by the final trimester, with the clearance reverting to its baseline value by three weeks after giving birth. In 69 pregnancies in which lamotrigine was used in monotherapy, Reisinger et al. (2013) found the mean apparent clearance of the drug in L per day was first trimester, 1.64 ± 0.82 [0.068 L/h]; second trimester, 2.53 ± 1.47 [0.105 L/h]; and third trimester, 2.09 ± 1.01 [0.087 L/h], as compared with a value of 0.87 ± 0.42 [0.036 L/h] when not pregnant. Polepally et al. (2014), on the basis of a population pharmacokinetic analysis, found that two sets of pregnant women existed in relation to their capacities to eliminate lamotrigine. The smaller group had only a small increase in clearance; in the larger group, the clearance increase was roughly an order of magnitude greater. They did not determine the different clearance mechanisms that were involved.
Pennell et al. (2004) inferred that an increased renal excretion of the drug might be responsible for the drug’s increased clearance during pregnancy. However, when Ohman et al. (2008) measured the ratio of the circulating concentrations of lamotrigine-2-N-glucuronide to lamotrigine in pregnancy and at 3 months postpartum, they found the ratio was 154 % higher during pregnancy. The subsequent evidence continues to be consistent with the maternal metabolism of lamotrigine being increased during pregnancy via enhanced glucuronidation . Reimers et al. (2011) found that the lamotrigine clearance was 118 % higher in the eighth month of pregnancy than in the second, and in the eighth month, the concentration ratio of lamotrigine-2N-glucuronide to lamotrigine in plasma was increased by 164 %. These workers also noted that, though plasma lamotrigine levels had begun to fall relative to drug dose in the second month of pregnancy, the plasma lamotrigine-2N-glucuronide to lamotrigine ratio was unchanged at that stage. They interpreted this finding in terms of plasma lamotrigine levels initially falling because of increased renal excretion of the drug, with the enhanced glucuronidation developing later. The increased glucuronidation of lamotrigine during pregnancy appears to correlate with the statement of Sabers (2008) that oestrogens induce UDP-glucuronosyl transferases and with the demonstration of Chen et al. (2009) that the enzyme induction is produced by 17-β-oestradiol .
The Foetus
In an ex vivo study, Myllynen et al. (2003) showed that lamotrigine crossed the perfused human placenta from the maternal to the foetal side readily and achieved a mean foetal–maternal ratio of 0.83 ± 0.41 at a maternal lamotrigine plasma concentration of 2.5 mg/L, and a ratio of 1.26 ± 0.20 at a maternal 10 mg/L plasma concentration of the drug.
Some measurements of the concentration of lamotrigine in umbilical cord venous blood, relative to the simultaneous concentration of the drug in maternal blood, are available. The values provide some indication of the likely concentrations of the drug to which the foetus would be exposed, at least in later pregnancy. Fotopoulou et al. (2009) found that the concentrations were virtually identical in maternal and umbilical vein blood. Kacirova et al. (2010) determined that the median infant to maternal plasma concentration ratio was 0.91 (range 0.40–1.38).
Breast Milk
There was interest in the concentration of lamotrigine in breast milk from a relatively early stage in the drug’s use in clinical practice. Rambeck et al. (1997) found a milk to maternal serum mean ratio of 0.56 ± 0.11 while they followed the parameter over 145 days of breast feeding in one patient. Tomson et al. (1997) found the ratio was 0.6 at 2 weeks postpartum, when the baby’s circulating lamotrigine concentration was 25 % of the maternal one. Ohman et al. (2000) cited the mean value for the ratio as 0.61 at a time when the baby’s lamotrigine concentrations had a mean value of about 30 % of that of the mother. De Haan et al. (2004) quoted a 0.54 value for the ratio, though only three women were studied, Newport et al. (2008) a mean value of 0.41, Fotopoulou et al. (2009) one of 0.59 and Clark et al. (2013) one of 0.33. Davanzo et al. (2013) reported a wide range of values for the parameter (0.57–1.47).
The Neonate
As mentioned immediately above, during breast feeding the baby’s circulating lamotrigine concentrations are about 25 % or 30 % of those in the mother, though Newport et al. (2008) cited a figure of 18.3 %. There is a report of an infant who became unacceptably sleepy while being breast fed by a mother who was taking lamotrigine. The baby, who had a circulating lamotrigine concentration of 4.87 mg/L, became alert again once breast feeding was ceased (Nordmo et al. 2009).
Published data for the elimination half-life of the drug in the neonate who is not breast fed have not been traced.
Topiramate
Topiramate ’s antiepileptic effectiveness was discovered after it was realised that the molecule, synthesised for a different purpose, possessed a degree of structural resemblance to that of acetazolamide , a drug that had some success in treating epilepsy but never became established in that role (Shank et al. 2000). Topiramate has proved a reasonably potent antiepileptic agent that has achieved moderately widespread use in recent years, at least in more affluent countries. The drug has subsequently found increasing employment in migraine prevention.
Chemistry
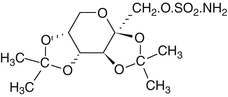
Chemically, topiramate is 2,3:4,5-bis-O-(1-methylethylidene)-α-D-fructopyranose sulphamate, a whitish monosaccharide derivative (MW 339.37, pKa value 8.61). It is moderately soluble in water and is marketed in solid dosage forms for oral use, mainly as tablets containing 25, 50, 100 and 200 mg of the active ingredient, and is also available as a sprinkle preparation.
Pharmacodynamics
Topiramate has several known mechanism of action at the molecular level. The role that each of these mechanisms plays in its antiepileptic, migraine -preventing and other actions is unclear. The drug inhibits voltage-sensitive Na+ and high-voltage-activated L-type Ca2+ ion channels , inhibits kainate-activated AMPA -type glutamate receptors and potentiates the actions of GABA in increasing Cl− flux at GABA receptors. It also is a relatively weak carbonic anhydrase inhibitor, and as already mentioned, a rather more potent inhibitor, acetazolamide , had previously been used for treating some forms of childhood epilepsy. Any or all of these mechanisms of actions might achieve an antiepileptic effect. However, the drug’s mechanism of action on Na+ channels is not identical with that shared by phenytoin , carbamazepine and lamotrigine . The mechanism through which topiramate prevents migraine is currently unexplained.
Clinically, the drug is capable of controlling all varieties of focal (partial) epilepsy and the generalised convulsive seizures of idiopathic, i.e. genetic, generalised epilepsies . It is also useful in preventing the drop attacks of the Lennox–Gastaut syndrome but does not seem able to prevent absence seizures occurring.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


