Podocyte proliferation and dedifferentiation are thought to play a role
Idiopathic most common form
Heterogeneous secondary forms
Infection: HIV, parvovirus B19, and many others
Medications: Interferon, bisphosphonates (pamidronate), calcineurin inhibitors, and others
Miscellaneous: Autoimmune disease, thrombotic microangiopathy, genetic diseases
Blacks disproportionately affected in idiopathic and HIV-associated forms
Nephrotic syndrome and nephrotic-range proteinuria
Renal dysfunction
Often refractory to steroid therapy
Rapid progression to renal failure
At least 1 glomerulus with
Global or segmental collapse
Overlying podocyte hyperplasia and hypertrophy
Tubulointerstitial fibrosis, cysts, and inflammation
Immunofluorescence: Segmental IgM and C3
Electron microscopy
Wrinkled glomerular basement membranes with capillary loop collapse
Segmental foot process effacement
FSGS, cellular variant
Crescentic glomerulonephritis
FSGS, not otherwise specified (NOS)
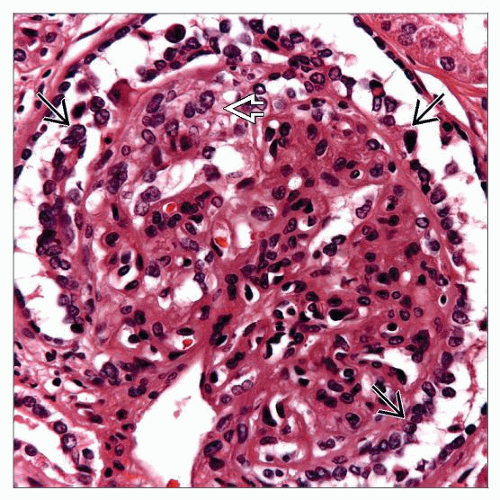 In collapsing glomerulopathy, glomerular capillary loops are not well defined 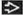 , and a rim of crowded, reactive podocytes is present , and a rim of crowded, reactive podocytes is present  . . |
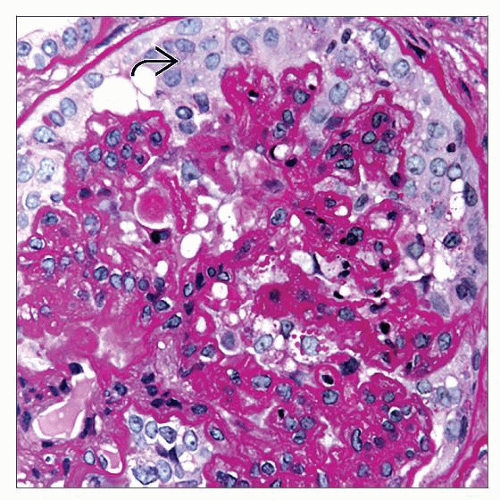 Podocyte proliferation in collapsing glomerulopathy can resemble a cellular crescent  . However, the cells are more rounded than the more spindle-shaped parietal epithelial cells typically seen in a crescent. . However, the cells are more rounded than the more spindle-shaped parietal epithelial cells typically seen in a crescent. |
Collapsing glomerulopathy (CG)
Focal segmental glomerulosclerosis (FSGS)
Primary FSGS, collapsing variant
Idiopathic collapsing FSGS
“Podocytopathy” defined pathologically by prominent capillary loop collapse, podocyte hypercellularity, and proliferation
Many different causes of collapsing podocyte phenotype
Target not limited to podocytes: Proximal tubular cells proliferate, and tubules are dilated
Idiopathic (usual)
Infection
Drugs
Vascular disease
Autoimmune disease
Malignancy
Genetic disorders
Cause unknown
Circulating permeability factor suspected but has limited experimental evidence
Genetic factors may contribute
Prevalent APOL1 in blacks has strong association with HIV-associated nephropathy and FSGS (and possibly with idiopathic CG)
Closely linked to MYH9 E1 haplotype (nonmuscle myosin heavy chain IIA)
CG is thought to be disorder resulting from podocyte proliferation and dedifferentiation
Normally, podocytes have low rate of turnover
WT-1 transcription factor inhibits proliferation
Podocytes in CG characteristically lose expression of WT-1 and increase expression of proteins involved in cell division (e.g., Ki-67)
Because of this, authors argue that CG may not be a form of FSGS since other forms of FSGS are considered to be due to loss of podocytes (podocytopenia)
FSGS consists of sclerosis (segmental solidification) of glomerulus and adhesions to Bowman capsule
In contrast, CG consists of pseudocrescent collapse of tuft with increased numbers of podocytes with few adhesions
CG also does not respond to typical therapies used for FSGS, suggesting different pathogenesis
Mitochondrial phenyltransferase-like protein mutations have been identified in kd/kd mice and associated with CG
May be involved in damage induced by some bisphosphonates
Ethnicity
Blacks are disproportionately affected (20-50x)
Drugs
None effective
Steroid therapy may be used, but disease may be refractory
Retinoic acid derivatives and inhibitors of cyclin-dependent kinesis may inhibit or reverse CG
Inhibits proliferation and promotes differentiation
Rapid progression to renal failure (6 months) is usual course of idiopathic CG, more rapid than other types of FSGS
Described as “malignant” FSGS variant when initially recognized in 1978 by Brown and colleagues
Other causes of CG also typically have rapid loss of renal function although improvement can occur with recovery from or removal of etiologic agent
Glomeruli
At least 1 glomerulus with global or segmental collapse and overlying podocyte hyperplasia and hypertrophy, according to Columbia Working Proposal
This is lowest possible threshold and has led to marked increase in diagnosis of CG
Podocytes with hypertrophy and hyperplasia
Urinary space may be filled with podocytes, forming pseudocrescents
Enlarged nuclei with open, vesicular chromatin and frequent nucleoli
Binucleate forms may be seen
Mitotic figures may rarely be seen
Protein resorption droplets may be seen in pseudocrescent podocytes
Glomerular basement membranes are wrinkled in areas of collapse
PAS and Jones methenamine silver stains are useful in highlighting basement membrane collapse
Mesangial and intracapillary matrix are not appreciably increased
Tubules
Tubular microcysts (in 40% of cases)
Proximal tubules dilated with proteinaceous casts, sometimes with “peripheral scalloping”
Proliferation of tubular cells
Enlarged hyperchromatic nuclei, mitotic figures, nucleoli, and focal apoptosis
Tubular atrophy/injury
Tubular epithelial simplification and flattening
Tubulitis can be present, often composed of neutrophilic tubulitis
Interstitium
Inflammation
Interstitial mononuclear inflammation can be prominent
Edema
Arteries/arterioles
Renal vessels may have changes of thrombotic microangiopathy if etiology involves TMA
IgM and C3 with segmental or global deposits in collapsed segments with less common deposits of C1q
IgG, IgA, and albumin in visceral epithelial protein resorption droplets
Tubules have epithelial protein resorption droplets containing plasma proteins (IgG, IgA, C3, albumin, and others)
Podocyte hypertrophy overlying areas of collapse
Foot processes are extensively effaced
Contain electron-dense protein resorption droplets, electron-lucent transport vesicles, and increased numbers of organelles, including prominent rough endoplasmic reticulum
Podocytes detached from glomerular basement membrane with interposition of newly formed extracellular matrix
Multiple layers of newly formed GBM between podocyte and original GBM
Actin cytoskeleton is disrupted, making cytoplasm appear open and pale
Podocytes become cuboidal
Glomerular basement membrane
Wrinkled GBM in areas of collapse
GBM not appreciably thickened
Absent electron-dense deposits except for small, rare paramesangial deposits
Glomerular endothelium
Absent tubuloreticular inclusions in all forms except for HIV-associated CG, interferon-mediated forms, and lupus-associated forms
Ki-67 (MIB-1), a proliferation marker, is positive in podocytes, indicating that they are engaged in proliferation
Normal podocytes have no or rare Ki-67(+) podocytes (< 1/glomerulus)
Podocyte differentiation markers are lost (e.g., WT-1, synaptopodin, podocin, podocalyxin, glomerular epithelial protein 1 [GLEPP1], C3b receptor, and CALLA [CD10]), suggesting that dedifferentiation plays a role in CG
CG lacks endocapillary hypercellularity seen in FSGS, cellular variant
Some authors consider cellular variant or “cellular lesion” to be same entity as CG
However, Columbia working proposal for FSGS classification (D’Agati et al) specifies that endocapillary hypercellularity is more evident in “cellular variant”
Endocapillary cells may even appear decreased in CG
Podocyte proliferation and hypercellularity absent
CG typically has less hyalinosis and adhesions to BC
Segmental and global sclerosis of usual type can be seen together with collapsing lesions, but collapsing features are considered to trump others and force classification to CG
Crescent cells generally do not have prominent reabsorption droplets
Hypertrophic podocytes of CG do not have spindled morphology of true crescents
True crescents also contain fibrin and matrix material not seen in pseudocrescents of CG
Necrotizing lesions in capillary tuft and glomerular basement membrane breaks
Prominent tubuloreticular structures in endothelial cells
May have mitochondrial abnormalities in tubules due to HAART
Evidence of thrombotic microangiopathy
Cholesterol emboli
Underlying glomerular disease
Lupus nephritis
Pathogenetic Classification of Collapsing Glomerulopathy | ||
Category | Cause | Comments |
Idiopathic | Possible circulating factor | Most common form, higher frequency in blacks; possibly associated with APOL1 variant |
Infection | ||
HIV | Widespread recognition in 1980s in HIV-associated nephropathy; higher frequency in blacks; associated with APOL1 variant | |
Parvovirus B19 | Viral antigen in podocytes | |
Leishmaniasis | ||
Loa loa | ||
Filariasis | ||
Tuberculosis | ||
Cytomegalovirus | ||
Campylobacter enteritis | ||
Drugs | ||
IV drug abuse (e.g., heroin) | Often associated with HIV | |
Bisphosphonates (e.g., pamidronate) | ||
Interferons-α and -β | ||
Valproic acid | ||
Calcineurin inhibitors | May originate through thrombotic microangiopathy-like state | |
Vascular | ||
Thrombotic microangiopathy | ||
Atheroemboli | ||
Autoimmune disease | ||
Guillain-Barré syndrome | ||
Systemic lupus erythematosus and lupus-like syndromes | ||
Mixed connective tissue disease | ||
Still disease | ||
Malignancy | ||
Leukemia/lymphoma | ||
Genetic | ||
CoQ2 nephropathy (co-enzyme Q2) | Described in Europeans | |
Mandibuloacral dysplasia (zinc metalloproteinase, ZMPSTE24) | Metalloproteinase is involved in post-translational cleavage of carboxy-terminal residues of farnesylated prelamin A | |
Action-myoclonus renal failure (SCARB2) | Myoclonic epilepsy associated with renal failure and preserved cognitive function | |
HIV-associated nephropathy, FSGS, and focal global glomerulosclerosis (hypertensive glomerular disease) | Associated with APOL1 variant that promotes resistance to trypanosomiasis | |
Familial CG | Unknown genes | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


