Coagulation Disorders
KEY CONCEPTS
![]() Hemophilia is an inherited bleeding disorder resulting from a congenital deficiency in factor VIII or IX.
Hemophilia is an inherited bleeding disorder resulting from a congenital deficiency in factor VIII or IX.
![]() The goal of therapy for hemophilia is to prevent bleeding episodes and their long-term complications and to arrest bleeding if it occurs.
The goal of therapy for hemophilia is to prevent bleeding episodes and their long-term complications and to arrest bleeding if it occurs.
![]() Recombinant factor concentrates usually are first-line treatment of hemophilia because they have the lowest risk of infection.
Recombinant factor concentrates usually are first-line treatment of hemophilia because they have the lowest risk of infection.
![]() Inhibitor formation is the most significant treatment complication in hemophilia. It is associated with significant morbidity and decreased quality of life.
Inhibitor formation is the most significant treatment complication in hemophilia. It is associated with significant morbidity and decreased quality of life.
![]() The goal of therapy for von Willebrand disease is to increase von Willebrand factor and factor VIII levels to prevent bleeding during surgery or arrest bleeding when it occurs.
The goal of therapy for von Willebrand disease is to increase von Willebrand factor and factor VIII levels to prevent bleeding during surgery or arrest bleeding when it occurs.
![]() Factor VIII concentrates that contain von Willebrand factor are the agents of choice for treatment of type 3 von Willebrand disease and some type 2 von Willebrand disease, and for serious bleeding in type 1 von Willebrand disease.
Factor VIII concentrates that contain von Willebrand factor are the agents of choice for treatment of type 3 von Willebrand disease and some type 2 von Willebrand disease, and for serious bleeding in type 1 von Willebrand disease.
![]() Desmopressin acetate often is effective for treatment of type 1 von Willebrand disease. It also may be effective for treatment of some forms of type 2 von Willebrand disease.
Desmopressin acetate often is effective for treatment of type 1 von Willebrand disease. It also may be effective for treatment of some forms of type 2 von Willebrand disease.
The coagulation system is intricately balanced and designed to stop bleeding at the site of vascular injury through complex interactions between the vascular endothelium, platelets, procoagulant proteins, anticoagulant proteins, and fibrinolytic proteins. Hemostasis stops bleeding at the site of vascular injury through the formation of an impermeable platelet and fibrin plug. Three key mechanisms facilitate hemostasis including vascular constriction, primary platelet plug formation (primary hemostasis), and clot propagation through fibrin formation (secondary hemostasis). Derangements in this finely tuned system can lead to either bleeding or thrombosis. Bleeding disorders are the result of either a coagulation factor defect, a quantitative or qualitative platelet defect, or enhanced fibrinolytic activity. The complex system regulating hemostasis is described in the pathophysiology section of Chapter 9.
COAGULATION FACTORS
Secondary hemostasis facilitates propagation and stabilization of the initial platelet plug formed in primary hemostasis through the formation of fibrin on the activated platelet surface. This step is initiated via the tissue factor pathway and is vital for adequate hemostasis. Coagulation factors circulate as inactive precursors (zymogens). Activation of these coagulation proteins entails a cascading series of proteolytic reactions (Fig. 9–2). At each step, a clotting factor undergoes limited proteolysis and becomes an active protease (designated by a lowercase “a,” as in Xa).
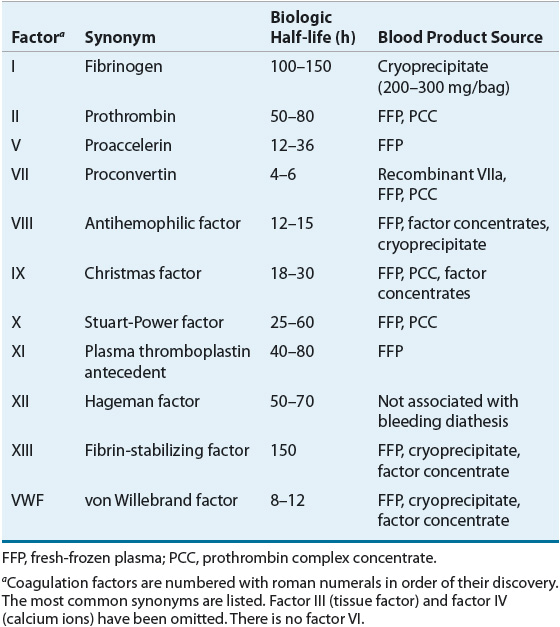
The coagulation factors can be divided into three groups on the basis of biochemical properties: vitamin K-dependent factors (II, VII, IX, and X), contact activation factors (XI and XII, prekallikrein, and high-molecular-weight kininogen), and thrombin-sensitive factors (V, VIII, XIII, and fibrinogen). Biologic half-life and blood product source varies by coagulation factor (Table 81–1).
TABLE 81-1 Blood Coagulation Factors
CLINICAL MANIFESTATIONS AND DIAGNOSIS
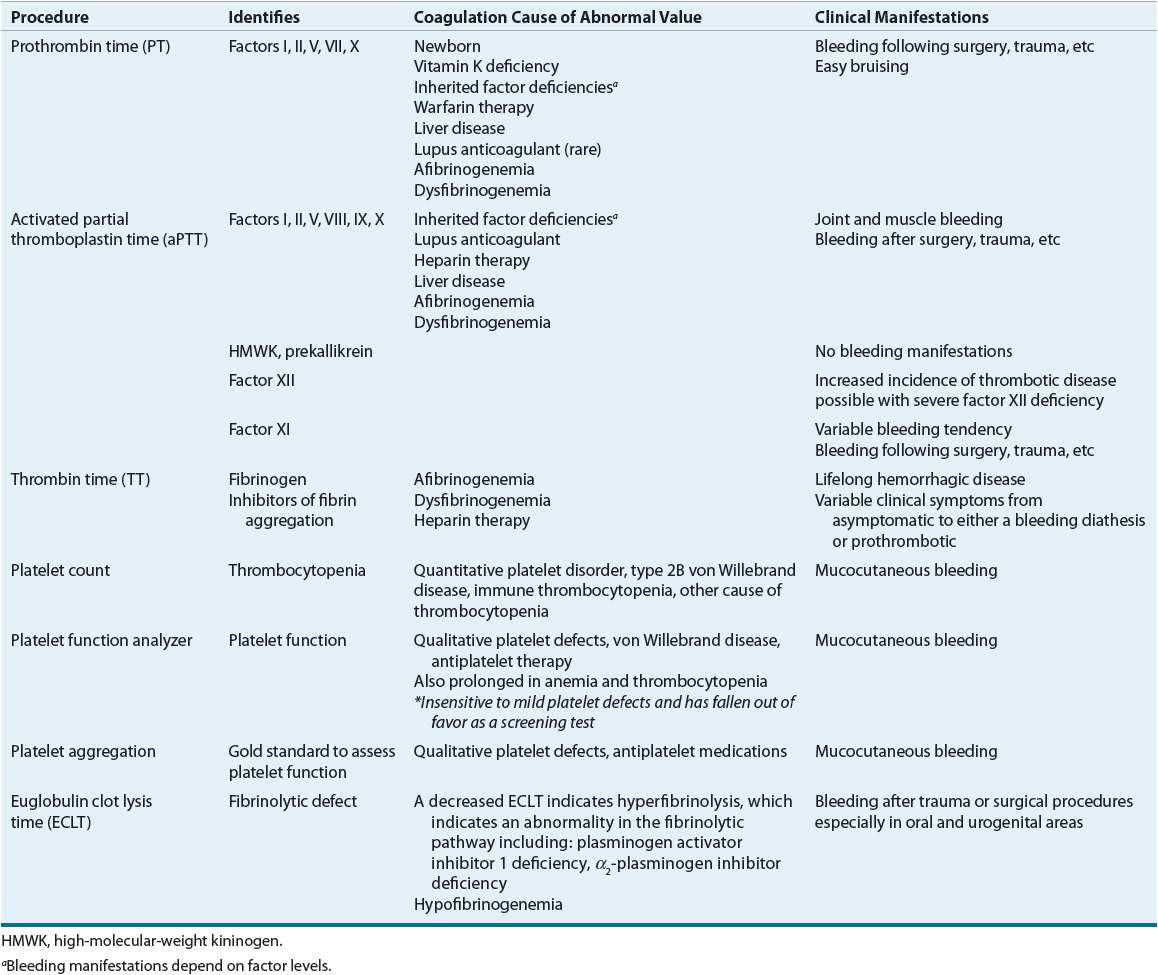
The diagnosis of coagulation disorders can be established from a detailed clinical history, physical examination, and laboratory test results. The clinical history should ascertain if there is a family history of bleeding or known bleeding disorders. Laboratory testing can distinguish bleeding disorders caused by defects in the coagulation pathways (Fig. 9–4), fibrinolytic pathways, or alterations in the number or function of platelets. Table 81–2 describes common coagulation tests.
TABLE 81-2 Laboratory Procedures
HEMOPHILIA
![]() Hemophilia is a bleeding disorder that results from a congenital deficiency in a plasma coagulation protein. Hemophilia A (classic hemophilia) is caused by a deficiency of factor VIII, whereas hemophilia B (Christmas disease) is caused by a deficiency of factor IX. The incidence of hemophilia is about 1 in 5,000 male births, 80% to 85% hemophilia A and 15% to 20% hemophilia B.1,2 There are no significant racial differences in the incidence of hemophilia.
Hemophilia is a bleeding disorder that results from a congenital deficiency in a plasma coagulation protein. Hemophilia A (classic hemophilia) is caused by a deficiency of factor VIII, whereas hemophilia B (Christmas disease) is caused by a deficiency of factor IX. The incidence of hemophilia is about 1 in 5,000 male births, 80% to 85% hemophilia A and 15% to 20% hemophilia B.1,2 There are no significant racial differences in the incidence of hemophilia.
About one-third of patients with hemophilia have a negative family history, presumably representing a spontaneous mutation. Both hemophilia A and hemophilia B are recessive X-linked diseases, which mean that the defective gene is located on the X chromosome. The disease primarily affects only males while females are carriers. Affected males have the abnormal allele on their X chromosome and no matching allele on their Y chromosome, their sons would be normal (assuming the mother is not a carrier) and their daughters would be obligatory carriers. Female carriers have one normal allele and therefore do not usually have a bleeding tendency. Although female carriers have lower factor VIII levels than females who are not carriers.3 Sons of a female carrier and a normal male have a 50% chance of having hemophilia and daughters have a 50% chance of being carriers. Thus, there is a “skipped generation” mode of inheritance in which the female carriers do not express the disease but can pass it on to the next male generation.
Hemophilia has been observed in a small number of females. It can occur if both factor VIII and IX genes are defective,4,5 if a female patient has only one X chromosome as in Turner syndrome, or if the normal X chromosome is excessively inactivated through a process called lyonization or highly skewed X inactivation.6–8
In 1984, researchers isolated and cloned the human factor VIII gene.9,10 It is a large gene, consisting of 186 kilobases (kb).11 More than 900 unique mutations in the factor VIII gene, including point mutations, deletions, and insertions, have been reported (http://hadb.org.uk/). Deletions and nonsense mutations are often associated with the more severe forms of factor VIII deficiency because no functional factor VIII is produced. In 1993, researchers identified an inversion in the factor VIII gene at intron 22 that accounts for about 45% of severe hemophilia A gene abnormalities.12 That discovery has greatly simplified carrier detection and prenatal diagnosis in families with this gene mutation. A more recently discovered inversion mutation involving intron 1 of the factor VIII gene accounts for an additional 5% of severe hemophilia mutations.13
The factor IX gene, cloned and sequenced in 1982, consists of only 34 kb and thus is significantly smaller than the factor VIII gene.14 Unlike the factor VIII gene in patients with severe hemophilia A, the factor IX gene in patients with hemophilia B has no predominant mutation. Direct gene mutation analysis is simpler in hemophilia B because of the smaller gene size, and to date more than 900 different mutations have been reported (http://kcl.ac.uk/ip/petergreen/haemBdatabase.html). Most of these mutations are single base-pair substitutions. About 3% of factor IX gene mutations are deletions or complex rearrangements, and the presence of these mutations is associated with a severe phenotype.11
Hemophilia B Leyden is a rare variant in which factor IX levels initially are low but rise at puberty. The mechanism underlying the pathogenesis of this disorder has been controversial. Some propose that the binding of the androgen receptor and other transcription factors are responsible.15,16 Other molecular mechanisms for age-related gene regulation has been recently discovered and implicated in factor IX Leyden.17 Identification of this genotype is clinically important because it confers a better prognosis.
Clinical Presentation
The characteristic bleeding manifestations of hemophilia include palpable ecchymoses, bleeding into joint spaces (hemarthroses), muscle hemorrhages, and excessive bleeding after surgery or trauma. The severity of clinical bleeding generally correlates with the degree of deficiency of either factor VIII or factor IX. Factor VIII and factor IX activity levels are measured in units per milliliter, with 1 unit/mL representing 100% of the factor found in 1 mL of normal plasma.18 Normal plasma levels range from 0.5 to 1.5 units/mL. Patients with less than 0.01 units/mL (1%) of either factor are classified as having severe hemophilia, those with 0.01 to 0.05 units/mL (1% to 5%) are moderate, and those with greater than 0.05 units/mL (5%) have mild hemophilia (Table 81–3).
TABLE 81-3 Laboratory and Clinical Manifestations of Hemophilia
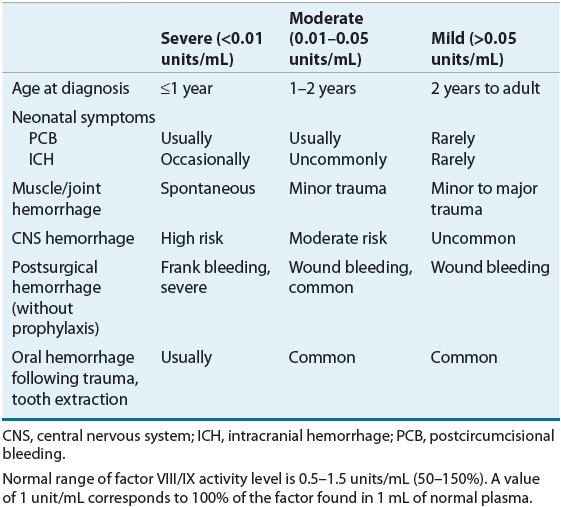
Patients with severe disease experience frequent spontaneous hemorrhages, whereas those with moderate disease have excessive bleeding following mild trauma and rarely experience spontaneous hemarthroses. Patients with mild hemophilia may have so few symptoms that their condition can be undiagnosed for many years, and they usually have excessive bleeding only after significant trauma or surgery. Disease severity does not always correlate with disease manifestations. Those with severe disease (less than 1% factor activity) may occasionally not display a severe phenotype, while some with milder forms of the disease may have more severe bleeding. Patients with hemophilia usually present with clinical manifestations after age 1 year, when they begin to walk and increase their risk of bleeding due to falling.
CLINICAL PRESENTATION Hemophilia
Diagnosis
The diagnosis of hemophilia should be considered in any male with unusual bleeding. A family history of bleeding is helpful in the diagnosis but is absent in up to 50% of patients with about one-third representing spontaneous mutations and the remaining secondary to an unrecognized family history.11 Brothers of patients with hemophilia should be screened; sisters should undergo carrier testing. Laboratory testing in patients with hemophilia will reveal an isolated prolonged partial thromboplastin time (PTT) and a decreased FVIII or FIX level.
Patients with severe hemophilia A should be tested for the common factor VIII gene inversions. In patients with severe hemophilia A who lack an inversion mutation or patients with moderate or mild hemophilia A, the gene can be sequenced to determine the exact mutation.19 Techniques for determining the genetic mutation in patients with hemophilia B are similar, but no predominant mutation like the factor VIII inversion has been found. The smaller size of the factor IX gene facilitates direct DNA mutational analysis.19
Advances in molecular genetic analysis have greatly improved the accuracy of carrier status evaluation. Thus, female relatives of patients with hemophilia who are at risk of being carriers should be tested. Carrier testing is simplified if the familial mutation has already been identified. Additionally, the appropriate factor level should be measured in female carriers to identify those with levels less than 0.3 units/mL (30%) who themselves might be at risk for bleeding.
Hemophilia can be diagnosed prenatally by chorionic villus sampling in gestational weeks 11 to 14 or by amniocentesis after 15 weeks’ gestation.20 These are invasive procedures with a 0.5% to 1% chance for pregnancy loss.20 A new noninvasive method uses cell-free fetal DNA that exists in maternal circulation to determine the sex of the fetus helping establish if more invasive testing is required for a male fetus.20 More recently this method was used to successfully identify hemophilia mutations in 12 subjects.21 At this time the method of using fetal DNA in maternal circulation to identify hemophilia mutations is still experimental and requires further validation.
TREATMENT
Hemophilia
Desired Outcomes
The comprehensive care of hemophilia requires a multidisciplinary approach. The patient is best managed in specialized centers with trained personnel and appropriate laboratory, radiologic, and pharmaceutical services. The healthcare team includes hematologists, orthopedic surgeons, nurses, physical therapists, dentists, genetic counselors, psychologists, pharmacists, case managers, and social workers. The goal for comprehensive hemophilia care is to preventing bleeding episodes and their long-term sequelae so that patients with hemophilia can live full, active, and productive lives.
![]() IV factor replacement therapy for the treatment or prevention of bleeding is the mainstay of treatment for hemophilia. Parents usually learn how to infuse factor concentrate to facilitate home treatment. Older children and adult patients learn self-administration. Home healthcare nursing support may be helpful, particularly for the youngest patients in whom venous access may be difficult. In the setting of poor venous access, central line placement may be indicated. Administration of factor at home is more convenient for families and allows for earlier treatment of acute bleeding episodes. However, serious bleeding episodes always require medical evaluation.
IV factor replacement therapy for the treatment or prevention of bleeding is the mainstay of treatment for hemophilia. Parents usually learn how to infuse factor concentrate to facilitate home treatment. Older children and adult patients learn self-administration. Home healthcare nursing support may be helpful, particularly for the youngest patients in whom venous access may be difficult. In the setting of poor venous access, central line placement may be indicated. Administration of factor at home is more convenient for families and allows for earlier treatment of acute bleeding episodes. However, serious bleeding episodes always require medical evaluation.
Patients with hemophilia should receive routine immunizations, including immunization against hepatitis B. Hepatitis A vaccine is also recommended for patients with hemophilia because of the risk (albeit small) of transmitting the causative agent through factor concentrates.22 Use of a small-gauge needle can prevent excessive bleeding. Many healthcare providers advocate subcutaneous rather than intramuscular immunizations to decrease the risk of intramuscular bleeding or hematoma formation, but there is a lack of evidence to support this route of administration.
A few special considerations apply to the perinatal care of male infants of hemophilia carriers. Intracranial or extracranial hemorrhage has been estimated to occur in 1% to 4% of newborns with hemophilia.23 Vacuum extraction and forceps delivery increase the risk of cranial bleeding. Elective cesarean section has not been shown to prevent intracranial bleeding. There is no clear consensus on the optimal mode of delivery or the use of prophylactic factor replacement in male infants of hemophilia carriers.23 Circumcision should be postponed until a diagnosis of hemophilia is excluded. Factor levels can be assayed from cord blood samples or from peripheral venipuncture. Arterial puncture should be avoided because of the risk of hematoma formation. If an infant has hemophilia, many clinicians recommend a screening head ultrasound to rule out an intracranial hemorrhage prior to discharge from the nursery.
History of Hemophilia Treatment
Therapy for hemophilia has undergone dramatic advances over the past few decades. Fifty years ago, administration of fresh-frozen plasma was the only available treatment. The introduction of cryoprecipitate in the early 1960s allowed more specific therapy for hemophilia A.24 Intermediate-purity factor VIII and IX plasma-derived concentrates became available in the 1970s.24 Plasma-derived factor concentrates are made from the donations of thousands of people. Contamination of plasma pools with hepatitis B, hepatitis C, and the human immunodeficiency virus (HIV) during the late 1970s and early 1980s resulted in transmission to a large portion of patients with hemophilia. Since the mid-1980s, plasma-derived concentrates have been manufactured with a variety of virus-inactivating techniques, including dry heat, pasteurization, and treatment with chemicals (e.g., solvent detergent mixtures).11 Since 1986, no transmission of HIV through factor concentrates to patients with hemophilia in the United States has been reported.11 Protein purification techniques, introduced in the 1990s, led to the production of high-purity plasma-derived concentrates with increased amounts of factor VIII or factor IX relative to the product’s total protein content. Recombinant factor VIII and then factor IX also became available in the 1990s.24 Significant improvements have also been made with recombinant products in limiting the risk of infectious transmission from albumin used to stabilize some of the products. Like plasma-derived products, these products use viral inactivation steps. With each subsequent generation of recombinant factor VIII products, the use of human proteins has been reduced.24
More recently, there has been significant progress in the development of long-acting factor VIII or IX products. Different methods have been utilized to prolong the half-life of either factor VIII or IX including pegylation, polysialic acid, albumin infusion, and Fc infusion.25 Clinical trials for factor VIII and FIX products that utilize either pegylation or fusion to an Fc receptor are ongoing.25
Hemophilia A
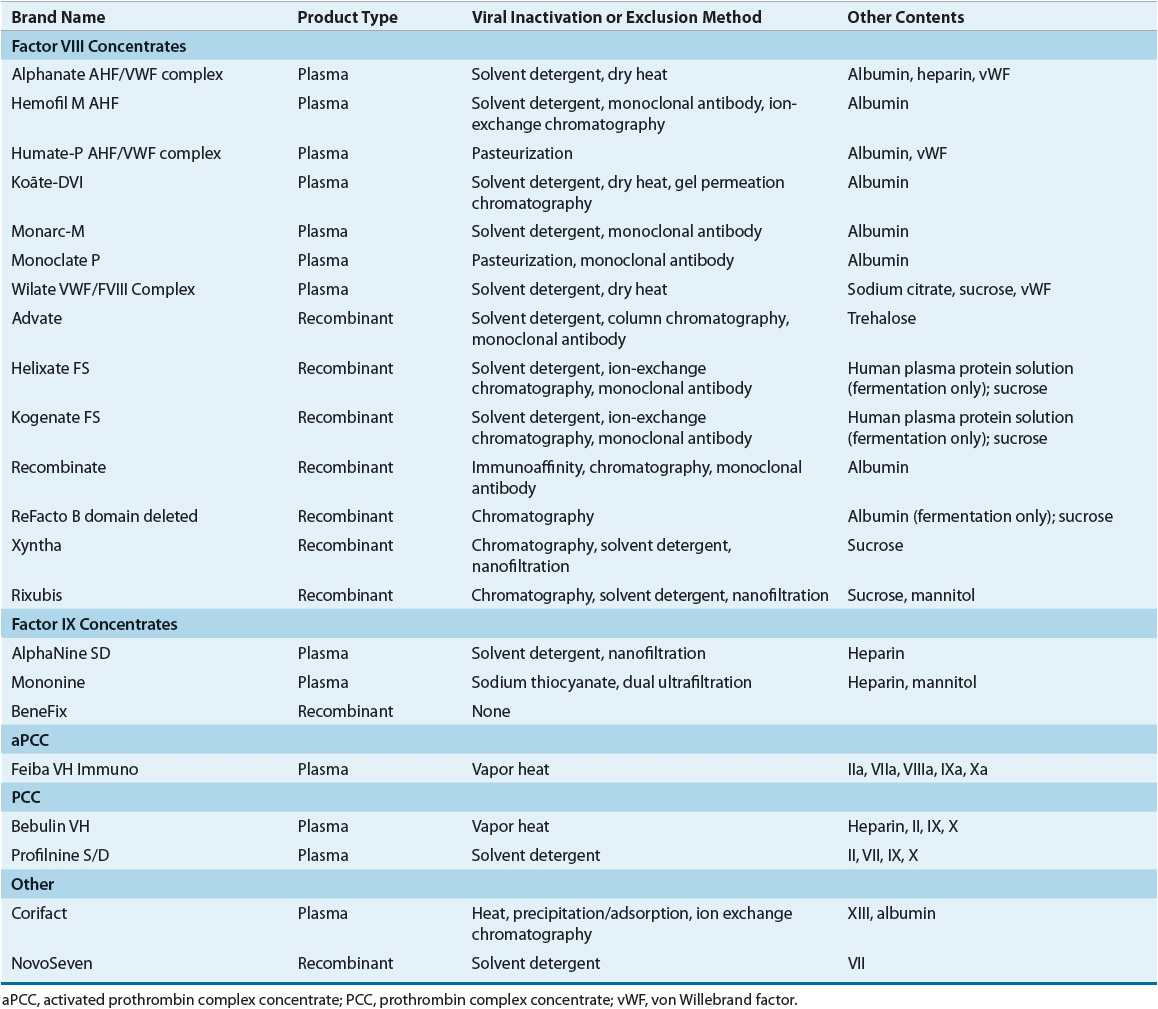
Table 81–4 summarizes the factor VIII products currently available in the United States. Most patients are treated with high-purity products. Products with the lowest risk of transmitting infectious disease should generally be used. Thus, recombinant products, when available, are generally used rather than plasma-derived products.
TABLE 81-4 Factor Concentrates
Recombinant Factor VIII
![]() Recombinant factor VIII is produced with recombinant DNA technology and is derived from cultured Chinese hamster ovary cells or baby hamster kidney cells transfected with the human factor VIII gene.11 Because it is not derived from blood donations, the risk of transmitting infections through administration of recombinant factor VIII is low and recombinant products are generally favored over plasma-derived products. A small risk of viral infection of the cell lines used to produce the clotting factor remains.26 Furthermore, human and/or animal proteins are utilized in the production process of some recombinant products.24 Therefore, these products have a theoretical risk of transmitting infection, although hepatitis and HIV infection have never been reported with their use.11 The presence of parvovirus B19 DNA has been reported in recombinant factor VIII products.27 First-generation recombinant factor VIII products contain human albumin as a stabilizing protein.11 Second-generation recombinant factor VIII products add sugar instead of human albumin as a stabilizer, but human albumin is utilized in the culture process. One second-generation product (ReFacto®) has deletion of the B domain of the factor VIII gene, yielding a smaller protein product.11 This B domain does not appear to be necessary for coagulation function. Third-generation recombinant factor VIII products contain no human protein either in the culture or in the stabilization processes.24
Recombinant factor VIII is produced with recombinant DNA technology and is derived from cultured Chinese hamster ovary cells or baby hamster kidney cells transfected with the human factor VIII gene.11 Because it is not derived from blood donations, the risk of transmitting infections through administration of recombinant factor VIII is low and recombinant products are generally favored over plasma-derived products. A small risk of viral infection of the cell lines used to produce the clotting factor remains.26 Furthermore, human and/or animal proteins are utilized in the production process of some recombinant products.24 Therefore, these products have a theoretical risk of transmitting infection, although hepatitis and HIV infection have never been reported with their use.11 The presence of parvovirus B19 DNA has been reported in recombinant factor VIII products.27 First-generation recombinant factor VIII products contain human albumin as a stabilizing protein.11 Second-generation recombinant factor VIII products add sugar instead of human albumin as a stabilizer, but human albumin is utilized in the culture process. One second-generation product (ReFacto®) has deletion of the B domain of the factor VIII gene, yielding a smaller protein product.11 This B domain does not appear to be necessary for coagulation function. Third-generation recombinant factor VIII products contain no human protein either in the culture or in the stabilization processes.24
Clinical trials have demonstrated that recombinant factor VIII products are comparable in effectiveness to plasma-derived products.11 The risk of patients with severe hemophilia A developing an inhibitory antibody to factor VIII with use of recombinant factor VIII is 25% to 32%.28 The risk of inhibitor formation has been reported to be higher in recombinant products as compared with plasma-derived products. In studies of previously untreated patients, the cumulative incidence of inhibitor formation was 10.3% for plasma-derived versus 28.7% for recombinant products.29 However, it is difficult to compare the cumulative incidence from different studies because of differences in patient population (e.g., heterogeneity in risk factors for inhibitor formation), study methodology, frequency of inhibitor testing, and length of follow-up.29 Two recent systematic reviews attempted to control for the heterogeneity in studies and were unable to demonstrate a difference in the risk of inhibitor formation.30,31 In the review by Iorio et al., most of the apparent difference in risk of inhibitor formation was explained by differences in study design, study period, testing frequency, and median follow-up. The source of factor concentrate (recombinant vs. plasma-derived) was not statistically significant.31 To address this very important clinical question, a prospective international randomized clinical trial (SIPPET—Survey of Inhibitors in Plasma Product Exposed Toddlers) is currently enrolling patients and is comparing inhibitor incidence in previously untreated patients exposed to either plasma or recombinant factor products.32
Plasma-Derived Factor VIII Products
Several different plasma-derived factor VIII products are available (Table 81–4). These products are derived from the pooled plasma of thousands of donors and therefore potentially can transmit infection. Donor screening, testing plasma pools for evidence of infection, viral reduction through purification steps, and viral inactivation procedures (e.g., dry heat, pasteurization, and solvent detergent treatment) have resulted in a safer product. No cases of HIV transmission from factor concentrates have been reported since 1986.11 However, isolated cases of hepatitis C infection with use of plasma-derived products have been reported.11 Additionally, outbreaks of hepatitis A viral infections associated with plasma-derived products, likely because solvent detergent treatment does not inactivate this nonenveloped virus, have been reported. Parvovirus has been reported to be present in both plasma-derived and recombinant factor VIII products.26,27 Finally, possible infection with as yet unidentified viruses that currently used methods would not inactivate remains a concern. In addition, Prion disease may be present in plasma-derived factor VIII products.26,33
Factor VIII concentrates can be classified according to their level of purity, which refers to the specific activity of factor VIII in the product. Cryoprecipitate is a low-purity product. Cryoprecipitate also contains von Willebrand factor, fibrinogen, and factor XIII. Current American Association of Blood Banks standards call for a minimum of 80 international units of factor VIII per cryoprecipitate pack.11 This product is no longer considered a primary treatment of factor VIII deficiency in countries where factor VIII concentrates are available because cryoprecipitate does not undergo a viral inactivation process. Intermediate-purity products have a specific factor VIII activity of 5 units/mg of protein and high-purity products have up to 2,000 units/mg of protein.11 Ultrahigh-purity plasma-derived products are prepared with monoclonal antibody purification steps and have a specific activity of 3,000 units/mg of protein prior to addition of albumin as a stabilizer.
Factor VIII Concentrate Replacement
Appropriate dosing of factor VIII concentrate depends on the half-life of the infused factor, the patient’s body weight, and the volume of distribution. The presence or absence of an inhibitory antibody to factor VIII and the titer of this antibody also influence treatment. Recovery studies, which measure the immediate postinfusion factor level, and survival studies, which assess the half-life of the factor, can establish patient-specific pharmacokinetics. The location and magnitude of the bleeding episode determine the percent correction to target as well as the duration of treatment. Serious or life-threatening bleeding requires peak factor levels of greater than 0.75 to 1 units/mL (75% to 100%); less severe bleeding may be treated with a goal of 0.3 to 0.5 units/mL (30% to 50%) peak plasma levels. Table 81–5 provides general guidelines for the management of bleeding in different locations.
TABLE 81-5 Guidelines for Factor Replacement Therapy for Hemorrhage in Hemophilia A and B

Factor VIII is a large molecule that remains in the intravascular space. Therefore, the plasma volume (about 50 mL/kg) can be used to estimate the volume of distribution. In general, each unit of factor VIII concentrate infused per kilogram of actual body weight yields a 2% rise in plasma factor VIII levels. The following equation can be used to calculate an initial dose of factor VIII:
Factor VIII (units) = (Desired level – Baseline level) × 0.5 × (Weight [in kilograms])
The baseline level usually is omitted from the equation when it is negligible compared to the desired level. The half-life of factor VIII ranges from 8 to 15 hours. It is generally necessary to administer 50% of the initial dose about every 12 hours to sustain the desired level of factor VIII. A single treatment may be adequate for minor bleeding, such as oral bleeding or slight muscle hemorrhages. However, because of the potential for long-term joint damage with hemarthroses, 2 or 3 days of treatment is often recommended for these bleeds. Serious bleeding episodes may require maintenance of 70% to 100% factor activity for 1 week or longer. As previously mentioned, factor VIII dosing depends on several variables, and each case must be considered individually. Individual pharmacokinetics may help guide treatment, particularly for serious bleeding episodes.
Alternatively, factor VIII can be administered as a continuous infusion when prolonged treatment is required (e.g., in the perioperative period or for serious bleeding episodes). Infusion rates ranging from 2 to 4 units/kg per hour usually are given in fixed-dose continuous infusion protocols, with the aim of maintaining a steady-state level of 60% to 100%.34,35 Administration of factor concentrate via continuous infusion may reduce factor requirements by 20% to 50% because unnecessarily high peaks of factor VIII that occur with bolus injections are avoided.35 A gradual decrease in factor VIII clearance during the first 5 to 6 days of treatment contributes to the lower factor concentrate requirements.35 Daily monitoring of factor level can help determine the appropriate rate of infusion.
Administration of factor VIII concentrate via continuous infusion has been shown to be safe and effective, and it may be more convenient than bolus therapy for hospitalized patients.34,35 The advantages of continuous infusion include maintenance of a steady-state plasma level with avoidance of potentially subtherapeutic trough levels and reduced cost associated with decreased factor requirements. A potential side effect with continuous infusion is thrombophlebitis at the delivery site. Concomitant infusion of saline or the addition of heparin (2 to 5 units/mL) to the infusion bag can minimize this risk.35 Bacterial contamination of the concentrate is another theoretical concern and preparation of the infusion bag should occur under sterile conditions (i.e., under laminar flow).35 Finally, concerns about the stability of the formulations appear to be unwarranted, as most high-purity factor VIII concentrates have been shown to remain stable for at least 7 days after reconstitution.35 Exposure of factor VIII to light for 10 hours after reconstitution can decrease activity by 30%.35 Therefore, it would be prudent to shield the container with foil wrap or an appropriate bag.
Other Pharmacologic Therapy
Treatment with desmopressin acetate often is adequate for minor bleeding episodes in patients with mild hemophilia A. A synthetic analog of the antidiuretic hormone vasopressin, desmopressin, causes release of von Willebrand factor and factor VIII from endogenous endothelial storage sites. It appears to be most effective in patients with higher baseline factor VIII levels (0.1 to 0.15 units/mL).36 The recommended dose of desmopressin is 0.3 mcg/kg diluted in 50 mL of normal saline and infused IV over 15 to 30 minutes.36 Patients with mild or moderate hemophilia A should undergo a desmopressin trial to determine their response to this medication. At least a twofold rise in factor VIII to a minimal level of 0.3 units/mL within 60 minutes is considered an adequate response.37 In adults with mild hemophilia A, the response rate to desmopressin has been reported to be 80% to 90%.37 Pediatric studies have reported a lower rate of response ranging from 40% to 47%.37 Furthermore, the pediatric response rate was related to age; some nonresponding children became responders at an older age.37
Infusion of desmopressin can be repeated daily for up to 2 to 3 days. Tachyphylaxis, an attenuated response with repeated dosing, may develop after that time. The factor increase after the second dose of desmopressin is about 30% lower than after the initial dose.36 Factor concentrate therapy may be necessary if the patient requires additional treatment. Factor levels should be measured to ensure that an adequate response has been achieved. Treatment with desmopressin will not result in hemostasis in patients who have severe hemophilia and those who are only marginally responsive. Desmopressin should not be used as primary therapy for life-threatening bleeding episodes such as intracranial hemorrhage or for major surgical procedures when a minimum and sustained factor VIII concentration of 0.7 to 1 units/mL is required.
Desmopressin can be administered intranasally via a concentrated nasal spray.36 It elicits a slower and less marked response, with a peak effect in 60 to 90 minutes after administration, which is somewhat longer than with IV administration.36,37 The dosage is one spray (150 mcg) for patients who weigh less than 50 kg and two sprays (300 mcg) for those who weigh more than 50 kg.37 The nasal spray may serve as an alternative to the IV formulation, especially in patients with mild bleeding episodes.
Few adverse effects are associated with desmopressin. The most commonly observed side effect is facial flushing.36 Less frequently reported side effects include mild headaches, increased heart rate, and decreased blood pressure. Thrombosis is a rare complication associated with desmopressin.37 Because of its antidiuretic effects, desmopressin has the potential to cause water retention, which may lead to severe hyponatremia. This may be a particular problem in children younger than 2 years, in whom hyponatremic seizures have been reported.36 Therefore, desmopressin should be used with caution in this age group.37 Patients with congestive heart failure may be at increased risk for developing hyponatremia with use of desmopressin.37 Fluid restriction for 24 hours after the desmopressin dose and monitoring of urine output are recommended with desmopressin administration.37
Antifibrinolytic therapy inhibits clot lysis and therefore is a useful adjunctive therapy for the treatment of hemophilia. Antifibrinolytic agents are particularly beneficial for treatment of oral bleeding because of a high concentration of fibrinolytic enzymes in saliva. Antifibrinolytic therapy can also be helpful as adjuvant therapy in GI bleeding, epistaxis, or menorrhagia. The two currently available antifibrinolytics include aminocaproic acid and tranexamic acid. Aminocaproic acid is given at a dosage of 100 mg/kg (maximum 6 g) every 6 hours and can be administered orally or IV.11 The dosage of tranexamic acid is 25 mg/kg (maximum 1.5 g) orally every 6 to 8 hours.11
Hemophilia B
Therapeutic options for hemophilia B have improved greatly over the past several years, first with the development of monoclonal antibody-purified plasma-derived products and then with the licensure of recombinant factor IX. Products currently available in the United States for treatment of hemophilia B are listed in Table 81–4.
Recombinant Factor IX
Recombinant factor IX was not available until 1998, which is 6 years after the first recombinant factor VIII product.38 Recombinant factor IX is produced in Chinese hamster ovary cells transfected with the factor IX gene. Blood and plasma products are not used to produce recombinant factor IX or to stabilize the final product; thus, recombinant factor IX has an excellent viral safety profile.11,38 Clinical trials have shown the product to be safe and efficacious in the treatment of acute bleeding episodes and in the management of bleeding associated with surgical procedures.11,38 Although the half-life of recombinant factor IX is similar to that of the plasma-derived products, recovery is about 30% lower.38 As a result, doses of recombinant factor IX concentrate must be higher than those of plasma-derived products to achieve equivalent plasma levels. Because individual pharmacokinetics may vary, recovery and survival studies should be performed to determine optimal treatment.11 Recombinant factor IX is considered the treatment of choice for hemophilia B.
Plasma-Derived Factor IX Products
High-purity factor IX plasma concentrates have been available in the United States since the early 1990s.11,38 These products are derived from plasma through biochemical purification and monoclonal immunoaffinity techniques. Other viral inactivation measures, such as solvent detergent or chemical treatment, are also used.
Before the high-purity products were approved for use, hemophilia B patients were treated with factor IX concentrates that also contained other vitamin K-dependent proteins (factors II, VII, and X), known as prothrombin complex concentrates (PCCs). These products contain small amounts of activated factors generated during processing, and their use has been associated with thrombotic complications, including deep-vein thrombosis, pulmonary embolism, myocardial infarction, and disseminated intravascular coagulation.11,38 The risk of such complications is highest in patients who are receiving high or repeated doses of PCCs, in those who have hepatic disease (the liver removes the activated factors from circulation), in neonates, and in patients who have experienced crush injuries or who are undergoing major surgery.11,38 Concomitant use of PCCs and antifibrinolytics should be avoided because of the risk for thrombosis.
Because of the lower purity of PCCs and their thrombogenic potential, these products are not first-line treatment of hemophilia B, although they are still used for treatment of patients with hemophilia A or B who have developed inhibitory antibodies against factor VIII or factor IX, respectively. High-purity factor IX concentrates have excellent efficacy in the treatment of bleeding episodes and in the control of bleeding associated with surgical procedures.38 Their viral safety profile has been reported to be excellent, and the risk of thromboembolic complications is low.38
Factor IX Concentrate Replacement
Factor IX is a relatively small protein. Unlike factor VIII, it is not limited to the intravascular space; it also passes into the extravascular compartment.38 This results in a volume of distribution that is about twice that of factor VIII. For plasma-derived factor IX concentrates, each unit of factor IX infused per kilogram of actual body weight yields about a 1% rise in the plasma level of factor IX (range, 0.67% to 1.28%).11 The following equation can be used to calculate the initial dose:
![]()
As with the factor VIII dose calculation, the baseline level term can be omitted from the formula if it is negligible compared to the desired level. Because recovery of recombinant factor IX is lower than that of the plasma-derived products, the following adjustment is made:
Pediatric dosing:
![]()
Adult dosing:
![]()
A recovery study to determine optimal dosing is recommended for patients who receive recombinant factor IX because of the wide interpatient variability in pharmacokinetics.
Because the half-life of factor IX is about 24 hours, dosing can be less frequent than with factor VIII. Table 81–5 provides general guidelines for dosing factor IX based on the site and severity of the bleeding episode.
Prophylactic Replacement Therapy
Traditionally, factor concentrates for hemophilia patients have been given on demand, as the bleeding episode occurs. However, recurrent joint bleeding can damage the joint and lead to the development of severe physical disability. Thus, it would be preferable to prevent bleeding episodes and avoid the resultant damage. Known as prophylactic factor replacement therapy, this approach consists of regular infusion of concentrate to maintain the deficient factor at a minimum of 0.01 units/mL (1%). In developed countries, prophylaxis for patients with severe hemophilia is considered standard of care. Patients with moderate hemophilia may sometimes require prophylaxis and it is rarely used in patients with mild hemophilia.
In effect, prophylactic replacement therapy converts severe hemophilia into a milder form of the disease. The rationale for this approach is that patients with moderate hemophilia rarely experience spontaneous hemarthroses, and they have a much lower incidence of chronic arthropathy. Two recent pediatric clinical trials have demonstrated the efficacy of prophylaxis in pediatric patients.39,40 The first pediatric randomized clinical trial comparing prophylaxis to enhanced episodic treatment to prevent joint disease in boys (age <30 months) with severe hemophilia demonstrated that prophylaxis prevented joint damage and decreased the frequency of joint and other hemorrhages.41 More recently, a European randomized clinical trial of prophylaxis in pediatric patients with hemophilia A confirmed the efficacy of prophylaxis in preventing bleeds and arthropathy.39 The efficacy of prophylaxis in adult patients with hemophilia is unclear and is the focus of ongoing clinical trials.
At this time, no consensus exists regarding the timing for the initiation of prophylaxis or the dosing schedule.25 A common regimen for patients with hemophilia A is 25 to 40 units/kg of factor VIII given every other day or three times per week. For hemophilia B, the usual dosage is 30 to 40 units/kg of factor IX given twice weekly because of the longer half-life of factor IX.38
Controversy exists regarding the ideal timing for the initiation of prophylaxis. Primary prophylaxis is regular replacement therapy started at a young age (usually before age 2 years), prior to the onset of joint bleeding.41 Secondary prophylaxis begins after significant joint bleeding has already occurred.41 In 2001, the Medical and Scientific Advisory Council of the National Hemophilia Foundation of the United States recommended primary prophylaxis beginning at age 1 to 2 years for children with severe hemophilia. The use of primary prophylaxis has many challenges and has not been widely accepted in the United States. Many institutions continue to use some form of secondary prophylaxis, in which prophylaxis is started after a pattern of bleeding has been established.
Several disadvantages are associated with primary prophylaxis. Perhaps most important is the high cost of prophylactic replacement therapy. Other issues to consider are the inconvenience to families and possible difficulties with compliance. Central venous lines may be necessary for frequent administration of factor concentrates, particularly in children younger than 2 years, who are at the age targeted for initiation of primary prophylaxis regimens. Potential complications of central venous access include surgical risks, infection, and catheter-related deep-vein thrombosis. Catheter-related infections are common in patients with hemophilia and have been reported to occur in up to 0.2 to 2/1,000 catheter days.42 Catheter-related infections appear to be even more common in hemophilia patients who have developed inhibitory antibodies.42 Finally, routine use of primary prophylaxis may initially overtreat some patients with severe hemophilia who do not have a severe clinical phenotype.



