Clostridium difficile was identified as the etiologic agent of antibiotic-associated pseudomembranous colitis (PMC) in 1978 (
1,
2) and is now recognized as the most important identifiable cause of healthcare-associated infectious diarrhea.
C. difficile may be the only pathogen sufficiently prevalent to warrant testing on a routine basis during the evaluation of healthcare-associated diarrhea
(3,
4). It is estimated that each case of healthcare-associated
C. difficile infection (CDI) costs between $10,212 and $13,675 and results in 3.0 to 6.4 excess hospital days
(5). Readmission for CDI costs $128,200 per hospital per year
(6). A conservative annual cost estimate for CDI in the United States is $3.2 billion
(5). Our present understanding of the pathogenesis of
C. difficile disease and rationale for preventive and interventive measures is supported by (a) observations on antimicrobial use in patients who acquire this pathogen, (b) potential infectious reservoirs, (c) modes of
C. difficile transmission, and (d) host risk factors.
PATHOGENESIS
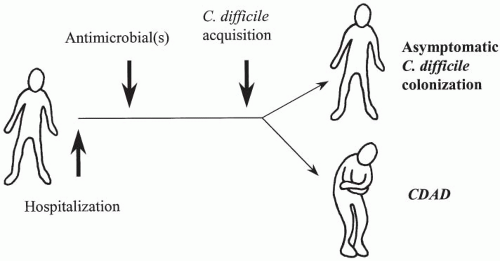
The manifestation of enteric disease due to C. difficile depends on at least three critical events: disruption of the normal colonic microflora, exposure to a toxigenic C. difficile strain, and the presence of one or more host factors. Epidemiologic evidence also supports the order of these events in that, with most cases, exposure to antimicrobials with subsequent compromise of host colonization resistance is followed by C. difficile acquisition from exogenous sources (rather than reactivation from an endogenous source).
The normal colonic flora provides a profound resistance to infection with
C. difficile. It appears that the host is susceptible to infection with this pathogen only after disruption of the colonic flora by antimicrobial therapy or by substances that act as antimicrobial surrogates such as antineoplastic agents
(7). The next critical event, exposure to toxigenic
C. difficile, occurs most often in hospitals and chronic care facilities, which serve as the main reservoirs for this infection. With the recognition that asymptomatic carriage of
C. difficile is very common among hospitalized patients, it might seem intuitive that these carriers are at high risk of subsequent CDI. However, a meta-analysis of four prospective studies that included 810 patients followed for 1,348 weeks with weekly surveillance cultures indicated that asymptomatic carriers were, conversely, at decreased risk of subsequent CDI (pooled risk difference -2.3% [95% confidence interval 0.3-4.3],
p .021)
(8,
9).
In our current hypothesis (
Fig. 37-1), a patient is admitted to the hospital and, although exposed intermittently to
C. difficile, is susceptible to colonization or disease only following antimicrobial therapy. The subsequent clinical outcome is determined shortly after acquisition (within a few days) and, like other enteric and infectious diseases, the majority of patients remain asymptomatic. These asymptomatic carriers are then at decreased risk for CDI when compared with noncolonized patients. Host risk factors comprise the third critical component to the pathogenesis of CDI and are discussed later in this chapter.
C. difficile elaborates two major toxins: toxin B, a potent cytotoxin; and toxin A, a potent enterotoxin that is also cytotoxic
(10). Although measurement of cytotoxicity in stool specimens is used to diagnose CDI, the enterotoxic effect of toxin A has been presumed to be critical in the pathogenesis of the disease. The early evidence implicating toxin A includes the observation that disease severity correlates more closely with toxin A production
in vivo (11) and that toxin A alone, but not toxin B, given intragastrically reproduces the pathology of
C. difficile cecitis in hamsters
(12). However, toxin B acts synergistically with toxin A in this model, and clinical data support virulence for variant strains that produce only toxin B, but not toxin A. Genetic knockout experiments indicate that toxin B, not toxin A, is the toxin essential for causing disease in the hamster model (
13). Furthermore, a monoclonal antibody directed at toxin A was ineffective at reducing CDI recurrence, whereas two monoclonal antibodies directed at toxins A and B were effective in reducing CDI recurrence in patients (
14,
15).
Toxin A is a unique enterotoxin unrelated to cholera toxin or the
Escherichia coli heat-labile toxin and causes extensive mucosal damage with hemorrhagic fluid response
(16). The receptor for toxin A involves a trisaccharide moiety, Gala1-3Galb1-4GlcNAc
(17), which is present on antigens within the brush border of human and hamster intestinal epithelium
(18). Subsequent toxic cellular events involve internalization of toxin A by receptor-mediated endocytosis and disruption of the cellular cytoskeleton. Toxin A (and B) induces glycosylation of small guanosine triphosphate-binding proteins that are important regulators of actin polymerization
(19).
Variant strains of
C. difficile that do not produce toxin A have been recovered from clinical specimens around the world. These toxin A−/B+ strains were initially recovered from asymptomatic children and were not thought to be pathogenic. Recently, a particular toxin A−/B+ variant that has a 1.8-kb deletion in the toxin A gene (referred to as toxinotype VIII, serogroup F, restriction endonuclease analysis [REA] group CF)
(20) has been recovered from multiple CDI cases, including a fatal case of PMC
(21). In addition, two well-documented hospital outbreaks with the A−/B+ variant
(22,
23) suggest that toxin B or some virulence determinant other than toxin A in these strains is sufficient to cause
C. difficile disease, consistent with the observations in hamsters using toxin A knockout
C. difficile strains (
13).
In addition to toxins A and B, some strains of
C. difficile (but not toxinotype VIII strains) also produce an adenosine diphosphate-ribosyltransferase (referred to as binary toxin)
(24). The role of binary toxin in the pathogenesis of CDI has not yet been determined, but during the first decade of the 21st century, there have been multiple outbreaks of a toxin-variant strain of
C. difficile that has caused severe CDI in North America, the United Kingdom, and Europe (
25,
26). It is a toxinotype III strain variously known as type NAP1 by pulse field, type 027 by polymerase chain reaction (PCR) ribotyping, and group BI by REA and is collectively termed BI/NAP1/027 (
25). The etiology of the high CDI rates and high severity of illness is not known, but the strains produce large amounts of toxins A and B
in vitro, produce binary toxin, and have a deletion in the
tcdC gene responsible for downregulation of toxin production
(27).
The other important aspect of
C. difficile pathogenesis lies within the apparent host resistance of some patients to
C. difficile disease. Infants in the first year of life frequently carry
C. difficile in high numbers with high levels of both toxins in their stools
(28), yet
C. difficile disease is rare in this group. Age-dependent expression of the epithelial cell receptor for toxin A may explain this apparent resistance in young infants
(29). Additionally, during outbreaks in adult settings, asymptomatic carriage is a more frequent outcome of CDI than is symptomatic infection
(8,
30). Disease manifestation and severity are not solely strain-specific phenomena
(31); although the mechanism of this apparent host resistance in adult asymptomatic fecal excretors is not completely known, host antibody response to toxin A has been one factor implicated (
32,
33).
CLINICAL DISEASE SPECTRUM
Although the most common clinical manifestation of CDI is diarrhea, the disease spectrum ranges from asymptomatic colonization or fecal excretion to PMC to septic shock with and without toxic megacolon, which may present with signs of an acute abdomen but without diarrhea
(34). As with other enteric infections, asymptomatic colonization is two to five times more common than clinical disease associated with
C. difficile (8,
30). Although colonized patients may carry epidemic strains responsible for illness in other patients, they are not at increased risk of CDI
(9), they are not at risk for subclinical protein-losing enteropathy
(35), and treatment of these patients with metronidazole or vancomycin is not advised
(36). Asymptomatic colonization is also very common in neonates, and it has been difficult to attribute any disease manifestation in neonates to
C. difficile. However, the pathogenic role of
C. difficile in children cannot be completely ignored, particularly in children over 1 year of age and especially in children with hypogammaglobulinemia
(37).
Diarrhea with or without demonstrable pseudomembranes in the colon is the most common manifestation of
C. difficile disease. Although
C. difficile is the most common recognized cause of antibiotic-associated diarrhea,
C. difficile accounts for only 15% to 25% of these cases
(38). CDI may occur during antimicrobial administration or several weeks after discontinuation of the antimicrobial. In a prospective study of clindamycin therapy, one-third of the patients developed diarrhea or colitis several days to 3 weeks after completion of clindamycin treatment
(39). This marked variability of time between onset of diarrhea and antimicrobial exposure also supports exogenous acquisition as the major source of CDI. The incubation period for diarrhea after acquisition of
C. difficile is less than 1 week, with a median onset of 2 days following acquisition
(8,
30). However, the incidence of CDI in the first 30 days following hospital discharge is very high, suggesting the possibility of either acquisition of the microorganism in the community following discharge or a longer incubation period (
40).
The severity and chronicity of diarrhea is also variable. In some cases, symptoms may be mild and respond to simply withdrawing the offending antimicrobial. CDI resolved spontaneously within 48 to 72 hours in 25% of patients in
one series
(41). More commonly, the diarrhea becomes chronic and severe if not diagnosed and treated with specific therapy. At presentation, symptoms may consist of only a few loose stools per day or multiple, large-volume, watery stools and signs of dehydration
(38). Stools may have mucus or evidence of occult blood but are rarely associated with visible blood
(42). Other findings commonly associated with CDI include abdominal pain (22%), ileus (21%), fever (28%), and leukocytosis (50%)
(42). CDI should be considered in any hospitalized patient with leukocytosis, particularly those with white blood cell counts >30,000 cells/mm3, even without the presence of diarrhea
(43). Complications of severe disease include dehydration, electrolyte imbalance, hypotension, hypoalbuminemia with anasarca, toxic megacolon, colonic perforation, and sepsis, and in 1% to 7% of cases, death can result (
26,
38). Higher mortality rates have been particularly noted in CDI caused by the epidemic BI/NAP1/027 strain (
26,
44). A characteristic of CDI is the high rate of clinical recurrence following successful therapy, which may result from either relapse with the same strain or reinfection with a new strain
(45,
46).
In distinction from antibiotic-associated diarrhea in general,
C. difficile is responsible for nearly all cases of PMC that have been reported since 1978
(38). The pseudomembranous intestinal lesions associated with
C. difficile (which are present in only about half the patients who have diarrhea and
C. difficile toxin in stool) have a characteristic gross and histologic appearance
(47). Early in the disease course, small (1-2-mm), raised, yellowish white plaques are noted, which may enlarge and coalesce
(48). These lesions, which are composed of fibrin, mucus, necrotic epithelial cells, and leukocytes, are restricted to the colon; therefore, this disease should be referred to as PMC rather than enterocolitis. Although PMC can be visualized by the sigmoidoscope in 90% of patients who have PMC, some patients have disease limited to the right colon, and the presentation may mimic appendicitis or Crohn disease
(49).
Fulminant
C. difficile colitis and toxic megacolon are less common manifestations, but important syndromes to recognize, as they are associated with a high mortality rate and frequently require surgical intervention
(50,
51). It is ironic that this most severe manifestation of CDI often occurs without diarrhea, and as a result, the diagnosis is frequently missed or delayed
(34). Risk factors for severe disease in one study included immunosuppression, prior CDI, and prior surgical procedures
(51). A rapidly increasing peripheral white blood cell count with a left shift may be an important clue to impending fulminant disease. A rising white blood cell count during medical treatment that is approaching 50,000/mm
3 or a lactic acidosis approaching 5 mmol/L are indication for colectomy in patients failing medical management
(52).
Extraintestinal CDIs are uncommon but include splenic abscess, bacteremia, wound infections, osteomyelitis, pleuritis, peritonitis, and urogenital tract infections
(53). As with other enteric infections, CDI has been associated with reactive arthritis
(54).