Chapter 17 Clinical development
present and future
Introduction
Bringing new drugs to the market is not only complex but also costly, time and resource consuming. Taking into account failure of drugs in development to make it to market, Di Masi (2003) estimated the costs to be around US$802 million and Adams and Brantner (2006) made an estimate of US$868 million but within a range of $500 million to $2 billion, depending on the indication pursued and the company performing the development. Of this total amount the clinical development accounts for just over half, i.e. about US$480 million.
In spite of heavy investment in research and development, new product approvals have been decreasing in the last 10 years (Woodcock and Woosley, 2008). Pipelines of large pharmaceutical companies have been declining and the productivity of large pharmaceutical companies in new drug development has been diminishing in an environment of increasing costs of clinical development and increasing risk aversion of companies, regulators and the general public. New strategies are needed to overcome this problem, both in the way companies source and develop new drugs and also in the way regulators approach the evaluation of efficacy and safety of new medicines.
In the United Sates the Food and Drug Administration (FDA) published a white paper in 2004 (FDA, 2004a) that identified that the current methods of drug development were partly behind the decline of new drug applications and has set up the Critical Path Initiative (FDA, 2004b) in order to address some of the problems of low productivity and high late-stage attrition rates, the idea being to encourage novel approaches to clinical development in trial design and measuring outcomes. The Innovative Medicines Initiative (EFPIA, 2008), underway in Europe, is also looking to encourage public and private collaboration with small and large enterprises and academia, to share knowledge, enhance the drug discovery and development process, reduce late-stage attrition and, ultimately, bring good medicines to patients in a cost- and time-effective manner.
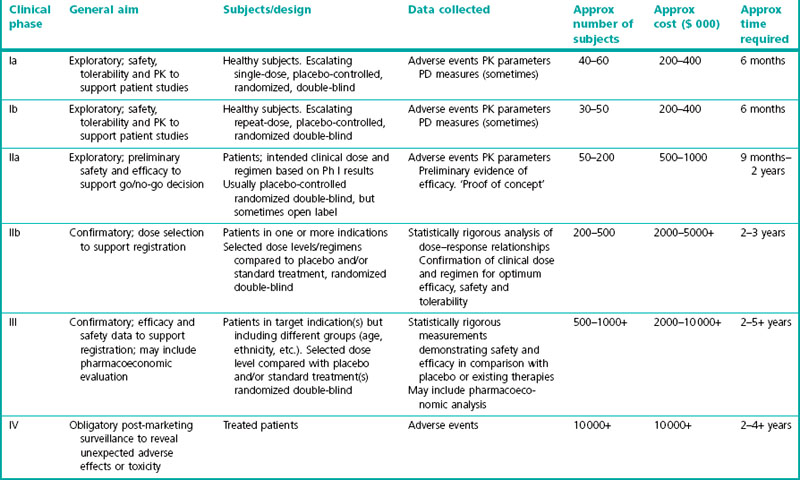
Clinical development phases
The conventional path of clinical development involves four phases:
• Phase I, pharmacokinetics, safety and tolerability and clinical pharmacology
• Phase IIa, exploratory efficacy
• Phase IIb, efficacy and dose range finding
• Phase III, pivotal efficacy and safety and larger population
A summary of these phases is given in Table 17.1.
Phase I – Clinical pharmacology
• First in man single ascending dose pharmacokinetics and safety
• Multiple ascending repeat dose pharmacokinetics and safety
• Bioavailability absolute and bioequivalence of new formulations
• Absorption distribution metabolism excretion in man (radiolabelled studies)
• Drug–drug interaction studies
• Safety pharmacology, e.g. thorough QT studies and abuse liability studies
• Elderly pharmacokinetics and safety
• Ethnic groups pharmacokinetics and safety
• Hepatic and renal impairment, pharmacokinetics and safety.
First in man, single ascending dose, pharmacokinetics and safety
Subjects
In most cases the subjects in first in man studies will be conducted in healthy young men usually aged 18–45 years. The idea behind this is to have a fairly homogeneous population in which to study the effects of the new drug and so limit variability, and also to have a population who will be more able to withstand any unexpected toxicity caused by the test drug. Healthy subjects are those who have no underlying diseases that could interfere with the conduct of the study or confound the interpretation of the safety or pharmacokinetic data. Criteria for inclusion into the study based upon medical history, physical examination, use of concomitant medications, alcohol, cigarettes and recreational drugs, as well as the results of blood testing 12-lead ECG, blood pressure heart rate are laid out in the study protocol. Male subjects are generally preferred, because at this early stage of development, reproductive toxicology testing in animals will not have been completed and the risk to the fetus of female subjects who might be pregnant or become pregnant shortly before or after the study, has not been characterized. Once the segment 2 reproductive toxicology has been completed (see Chapter 15), female subjects of non-childbearing potential, i.e. post-menopausal, surgically sterilized, sexually abstinent or using effective methods of contraception, may be included in European studies. In the USA, female subjects may be included prior to reproductive toxicology data being available if they are of non-childbearing potential. In some cases it is not appropriate to use healthy young men, for example, studies of female hormone products or products for oncology, and in these cases the subjects will be selected from the appropriate population.
Design
The choice of doses to be administered in the SAD trials should be based on the highest dose at which no adverse effects were seen in the most sensitive species tested in toxicology studies (the no observed adverse effect level, or NOAEL; see Chapter 15) and on the nature of the toxicity observed. The starting dose should be at least 10-fold lower than the NOAEL, but specific guidelines exist for the accurate determination of the starting dose on a case-by-case basis (FDA; http:www.fda.gov/cber/gdlns/dose.htm).
Typical dose escalation schedules from a starting dose of X are:
Outcome measures
In the SAD study the principal outcome measures are safety, tolerability and pharmacokinetics.
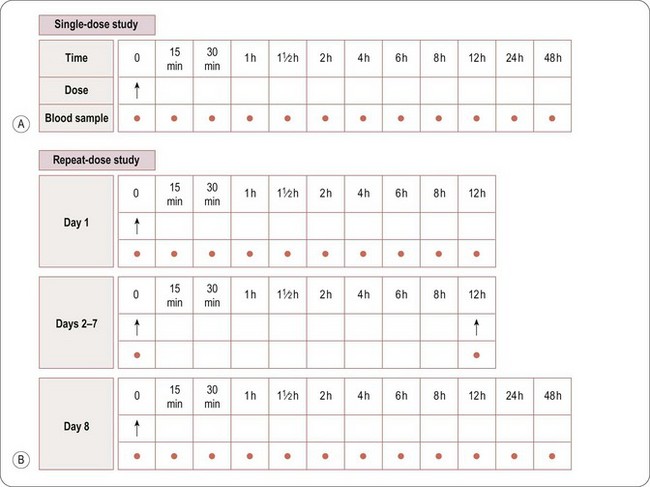
Whereas assessment of safety and tolerability is the primary objective, pharmacokinetic evaluation is the secondary objective of a SAD study. Blood samples will normally be taken before dosing and at specified intervals after dosing to measure the amount of drug in the blood or plasma at various time points after each dose. A typical sampling schedule is shown in Figure 17.1A. In addition, urine and/or faeces may be collected to measure the excretion of the drug via the kidneys and/or liver (in bile). The results enable the rate of absorption, metabolism and excretion of the drug to be explored, and the PK parameters to be estimated (see also Chapter 10).
The plasma or serum PK parameters usually derived are:
• Cmax: peak drug and/or metabolite(s) concentration
• Tmax: time to peak drug and/or metabolite(s) concentration
• AUC0–∞: area under the concentration–time curve of the drug and/or metabolite(s), extrapolated to infinity
• AUC0-T: area under the concentration–time curve of the drug and/or metabolite(s), calculated to a specific time point T
• t1/2: time taken for levels of drug and/or metabolite(s) to decrease by half (a measure of the rate of elimination of the drug from plasma).
Other PK parameters may also be determined, including:
• VD: volume of distribution of drug and/or metabolite(s)
• Cl: clearance of drug and/or metabolite, i.e. the volume of plasma/serum cleared of drug and/or metabolite(s) per unit time, e.g. mL/min, L/h
• MRT: mean residence time, i.e. the average time a drug molecule remains in the body after rapid i.v. injection.
Specialist pharmacokineticists perform the calculation of these parameters.
Multiple ascending repeat-dose studies
Outcome measures
The general requirements and procedures for MAD studies are the same as those for Phase I SAD. A typical Phase MAD blood sampling schedule is shown in Figure 17.1B. Blood samples for PK profiling will generally be taken on the first and last days of dosing, with additional single samples taken immediately before the first morning dose each day, to measure the levels of drug remaining in the blood immediately before the next dose is administered, i.e. the trough level.
Drug–drug interaction studies
The objective of drug–drug interaction studies (DDI) is to determine whether the test drug’s efficacy, safety or pharmacokinetics will be altered if it is given with other drugs that the target patient population may also be taking. The timing of drug interaction studies depends partly on the importance of understanding interactions prior to treating patients. Some DDIs may need to be performed prior to Phase IIa if the patient population in the study cannot be excluded from taking concomitant medications that might interact with the test drug. Otherwise DDIs can be conducted later in the development programme when more is known about the target treatment population and the efficacy and safety of the new drug. The choice of which DDI studies to perform is based on the following: the metabolism of the new drug (for example if it inhibits, induces or is metabolized by certain cytochrome p450 enzymes – see Chapter 10); the pharmacodynamic action of the drug; and which drugs the target population may be taking concomitantly. Drug interactions can occur on a metabolic level, so that the exposure to a certain dose may be changed by a drug interaction or on a pharmacodynamic level so that pharmacological effects may be increased or diminished by the interacting drug. It is important to known whether co-administration of the relevant drugs leads to a change in exposure or a change in pharmacodynamic effect of either the test drug or the interacting drug.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


