3
Clinical and Environmental Toxicity
Chapter 3 Clinical and Environmental Toxicity is a compilation of Chapter 4, Drug Toxicity and Poisoning and Chapter 67 Environmental Toxicology: Carcinogens and Heavy Metals from Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. The specific pharmacology (including the mechanisms of action) of drugs mentioned in Chapter 3 is discussed in previous or subsequent chapters. The Mechanisms of Action and the Clinical Summary Tables are limited to the commonly used antidotes, toxic metals, and the metal chelators that are used therapeutically to treat heavy metal poisoning. In addition to the material presented here, the 12th Edition includes:
• A detailed description of drug toxicity testing in animals
• A detailed description of drug safety testing and clinical trials in humans
• A list of important resources for information related to drug toxicity and poisoning
• A description of various methods of decontaminating a poisoned patient
• A discussion of environmental risk assessment and risk management
• A detailed discussion of carcinogenesis and chemoprevention
• A discussion of the principles of the treatment of metal exposure
LEARNING OBJECTIVES
 Understand the dose-response relationship of drugs and how it is used to quantify efficacy and toxicity.
Understand the dose-response relationship of drugs and how it is used to quantify efficacy and toxicity.
 Understand how drugs are tested for safety and efficacy in humans.
Understand how drugs are tested for safety and efficacy in humans.
 Know the different types of therapeutic drug toxicity.
Know the different types of therapeutic drug toxicity.
 Describe the different types of drug-drug interactions and know how they apply to drug therapy.
Describe the different types of drug-drug interactions and know how they apply to drug therapy.
 Know the principles of managing a drug poisoning including the safe use of specific antidotes.
Know the principles of managing a drug poisoning including the safe use of specific antidotes.
 Know the toxicity of acute and chronic heavy metal exposure.
Know the toxicity of acute and chronic heavy metal exposure.
 Understand the mechanisms of action and therapeutic uses of metal chelators for the treatment of acute metal exposure.
Understand the mechanisms of action and therapeutic uses of metal chelators for the treatment of acute metal exposure.
• Acetylcysteine (MUCOMYST)
• Aflatoxin B1
• Arsenic
• Atropine
• Benzo[a]pyrene
• Cadmium
• Chromium
• Deferasirox (EXJADE)
• Deferoxamine (DESFERAL)
• Dimercaprol
• Dimercaptosuccinic acid (DMSA), succimer (CHEMET)
• Diphenhydramine (BENEDRYL)
• EDTA CaNa2
• Ethanol
• Flumazenil
• Fomepizole
• Lead
• Mercury
• Naloxone (NARCAN)
• Penicillamine
• Physostigmine
• Pralidoxime chloride (2-PAM)
• Sodium 2,3-dimercaptopropane sulfonate (DMPS)—not approved by FDA, but approved for use in Germany
a A complete list of antidotes is shown in Table 3-1; b examples of important carcinogens are shown in Table 3-2; c a list of toxic metals with frequent environmental or occupational exposure is shown in Table 3-3.
TABLE 3-1 Some Common Antidotes and Their Indications
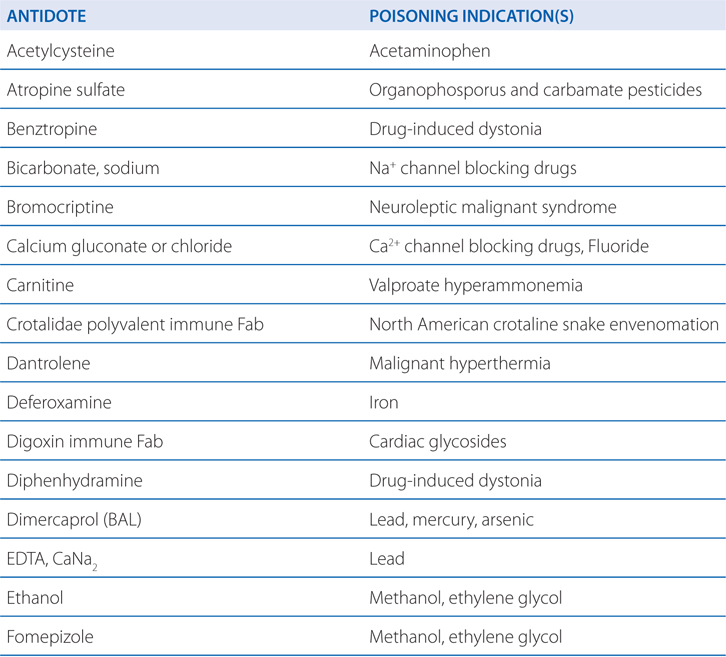
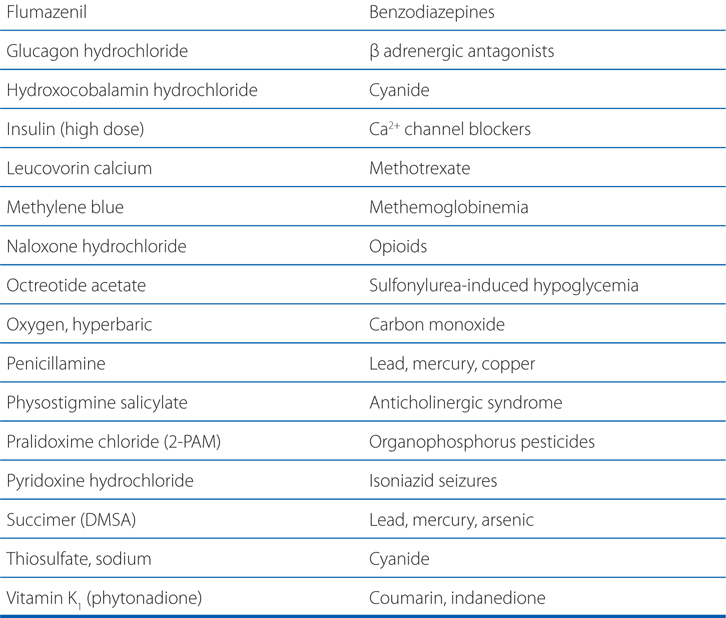
TABLE 3-2 Examples of Important Carcinogensa
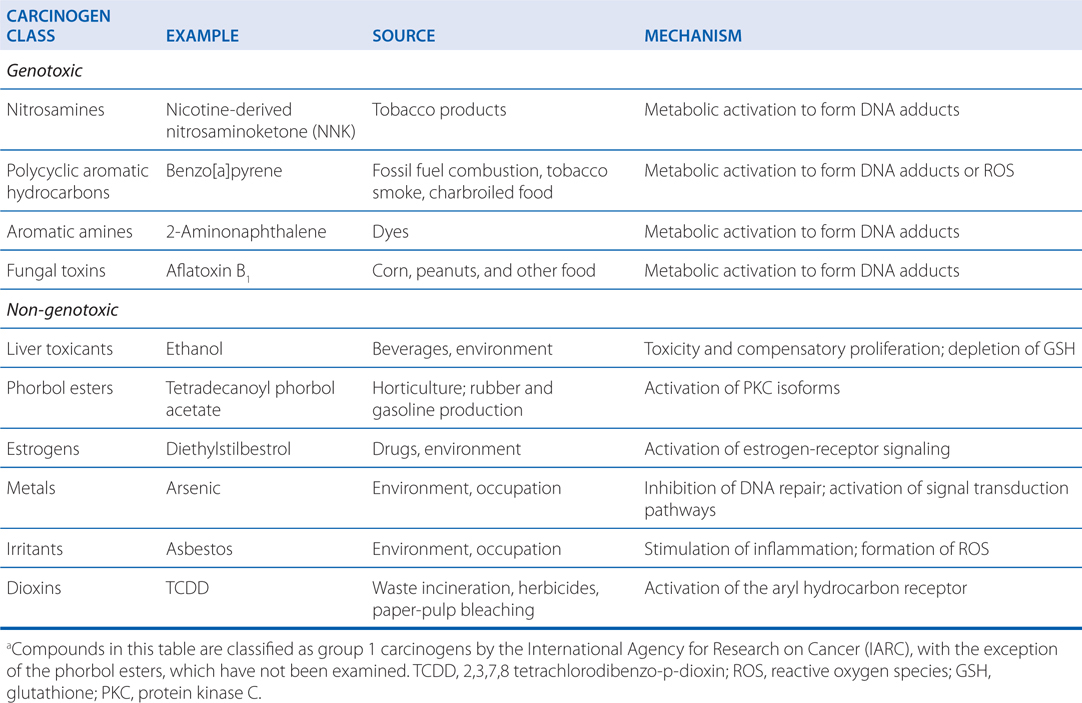
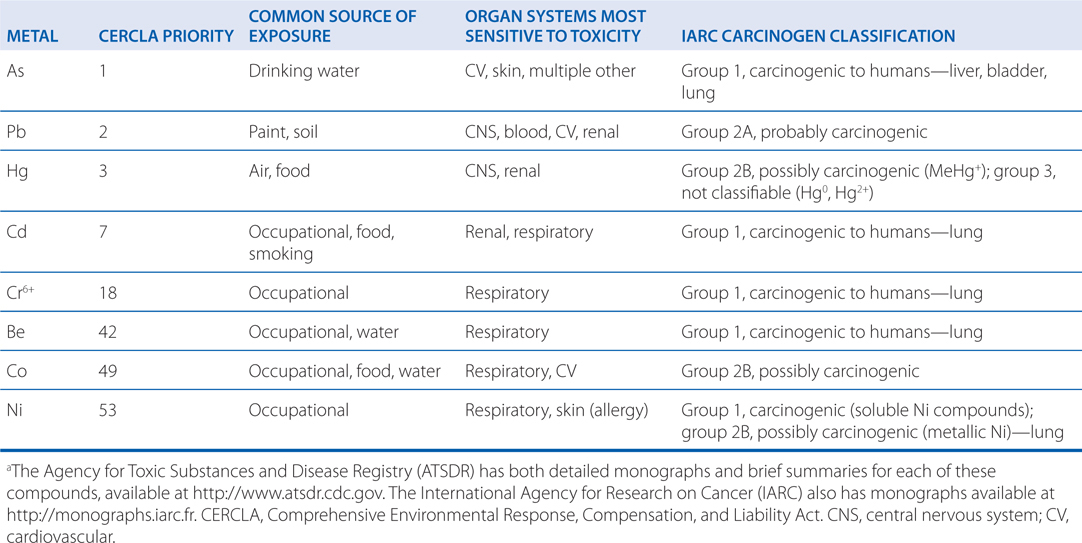
TABLE 3-3 Toxic Metals with Frequent Environmental or Occupational Exposurea
MECHANISMS OF ACTION OR TOXICITY OF SELECT ANTIDOTES, CARCINOGENS, AND HEAVY METALS
A new chemical is proposed for the treatment of hypertension.
a. Describe how the effects of the chemical are initially quantified?
Evaluation of the dose-response or the dose-effect relationship is crucially important to toxicologists. There is a graded dose-response relationship in an individual and a quantal dose-response relationship in the population (see Chapters 1 and 2). Graded doses of a drug given to an individual usually result in a greater magnitude of response as the dose is increased. In a quantal dose-response relationship, the percentage of the population affected increases as the dose is raised; the relationship is quantal in that the effect is specified to be either present or absent in a given individual (see Figure 3-1). This quantal dose-response phenomenon is extremely important in toxicology and is used to determine the median lethal dose (LD50) of drugs and other chemicals. The LD50 of a compound is determined experimentally, usually by administration of the chemical to mice or rats (orally or intraperitoneally) at several doses in the lethal range.
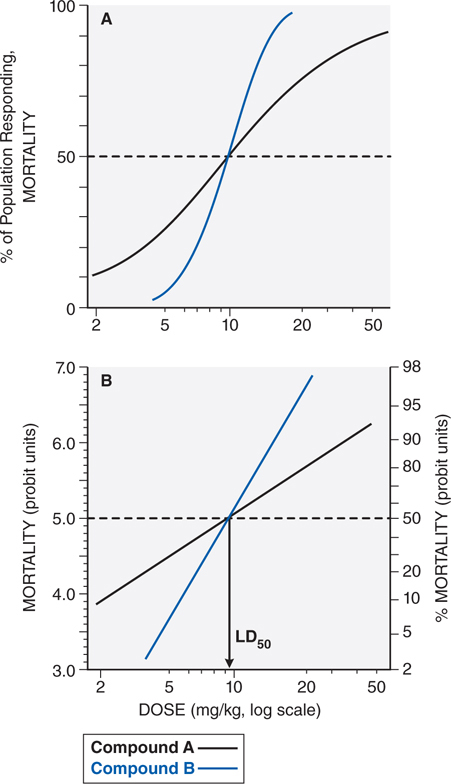
FIGURE 3-1 Dose-response relationships. A. The toxic response to a chemical is evaluated at several doses in the toxic or lethal range. The midpoint of the curve representing percent of population responding (response here is death) versus dose (log scale) represents the LD50, or the concentration of drug that is lethal in 50% of the population. B. A linear transformation of the data in panel A is provided by plotting the log of the dose administered versus the percent of the population mortality, in probit units. For a discussion of probit units, see Chapter 4 in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition.
b. Describe how the chemical’s toxicity and “therapeutic index” are determined?
Figure 3-2 illustrates the relationship between a quantal dose-response curve for the therapeutic effect of a drug to generate a median effective dose (ED50), the concentration of drug at which 50% of the population will have the desired response, and a quantal dose-response curve for lethality by the same agent. These 2 curves can be used to generate a therapeutic index (TI), which quantifies the relative safety of a drug.
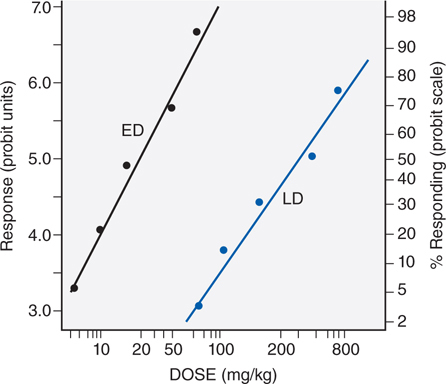
FIGURE 3-2 Comparison of effective dose (ED), and lethal dose (LD). For a discussion of probit units, see Chapter 4 in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. Note that the abscissa is a logarithmic scale.
TI = LD50/ED50
Drugs show a wide range of TI, from 1 to 2 to more than 100. Drugs with a low TI must be administered with caution. Agents that fall into this category include the cardiac glycoside digitalis (see Chapter 17) and cancer chemotherapeutic agents (see Chapters 45 and 46). Agents with very high TI are extremely safe and include some antibiotics (eg, penicillin) (see Chapter 39), unless there is a known allergic response.
GUIDELINES FOR THE STUDY OF NEW DRUGS IN HUMANS

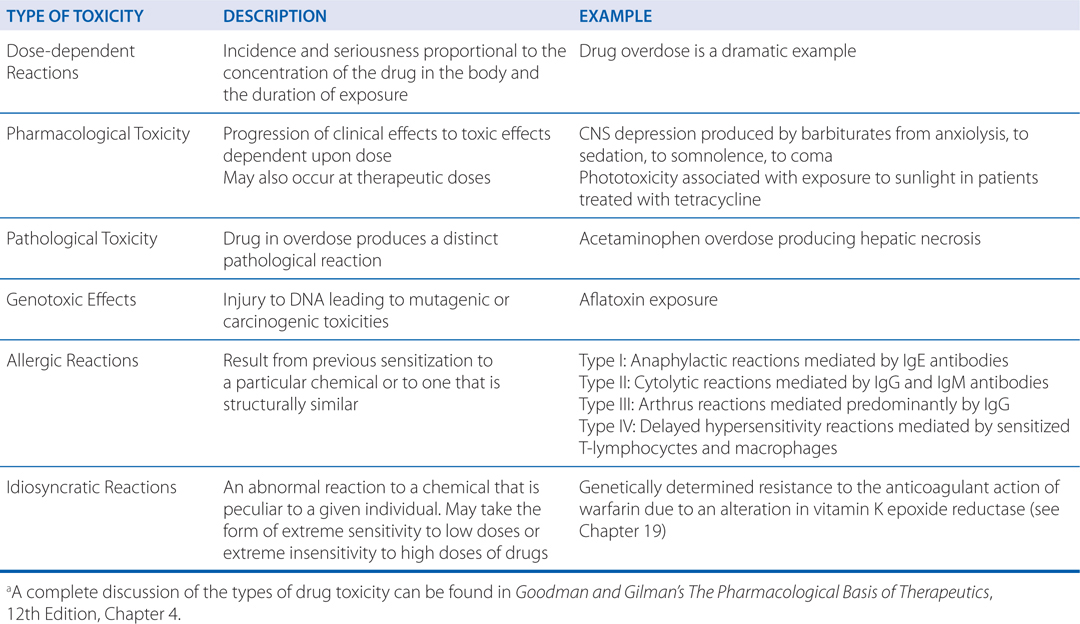
TYPES OF THERAPEUTIC DRUG TOXICITYa
A 35-year-old woman is being treated for esophageal reflux with a drug that has been recently approved by the Food and Drug Administration (FDA). After 2 months of therapy she is feeling better and nightly episodes of reflux are diminished, but her physician calls to tell her that the drug is being recalled because of a problem with cardiac arrhythmia, and a new drug will be prescribed.
a. What is the process of drug approval by the FDA?
Fewer than one-third of the drugs tested in clinical trials reach the marketplace. Federal law in the United States and ethical considerations require that the study of new drugs in humans be conducted in accordance with stringent guidelines. See Side Bar GUIDELINES FOR THE STUDY OF NEW DRUGS IN HUMANS.
b. What are the different types of therapeutic drug toxicity?
In therapeutics, a drug typically produces numerous effects, but usually only 1 is sought as the primary goal of treatment; most of the other effects are undesirable effects of that drug for that therapeutic indication (see Figure 3-3). Side effects of drugs usually are bothersome but not deleterious nor do they always necessitate the discontinuation of the drug; they include effects such as dry mouth occurring with tricyclic antidepressant therapy. Other undesirable effects may be characterized as toxic effects (see Side Bar TYPES OF THERAPEUTIC DRUG TOXICITY).
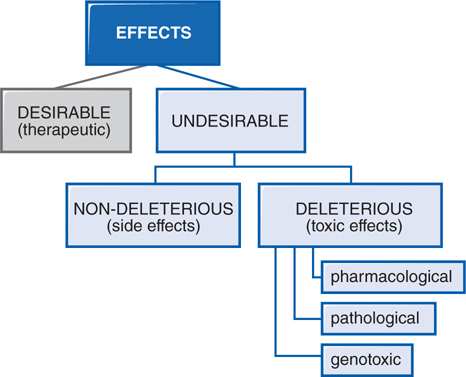
FIGURE 3-3 Spectrum of the effects of pharmaceuticals.
c. Why can a drug approved by the FDA for use in humans and still cause serious toxicity?
Some toxicities of pharmaceuticals can be predicted based upon their known pharmacological mechanism; however, it is often not until the postmarketing period that the therapeutic toxicity profile of a drug becomes fully appreciated. In the United States, the approval system for new drugs typically uses only 500 to 3000 exposed subjects. Such a system is likely to identify toxicities occurring in 1% or more of patients receiving a drug. The Adverse Event Reporting System of the FDA relies upon 2 signals to detect rare drug events. First, the FDA requires (Code of Federal Regulations, Title 21, Volume 5, Section 314.80) drug manufacturers to perform postmarketing surveillance of prescription drugs, and similar regulations exist for nonprescription products. Second, the FDA operates a voluntary reporting system MedWatch, available to both health professionals and consumers. Hospitals may also support adverse drug event committees to investigate potential adverse drug events, and these investigations may be made available to industry and the government. Unfortunately, any national dataset will significantly underestimate the morbidity and mortality attributable to adverse drug events because of underreporting and because it is difficult to estimate the denominator of total patient exposures for each event reported once a drug is available on the open market.
As an example, postmarketing surveillance identified the toxicity associated with the serotonin receptor-modulating drug cisapride. This drug was known to enhance GI motility and was marketed in the United States as a treatment for gastroesophageal reflux. Postmarketing surveillance revealed that cisapride was associated with prolongation of the QT interval and predisposition to ventricular arrhythmia. It was withdrawn from the market, and subsequent case-control studies demonstrated increased risk of arrhythmia. Cisapride is now limited in its distribution through an investigational access program managed by the manufacturer. Thus, the determination of drug toxicity extends beyond the stages of drug development and approval.

A 38-year-old man is being treated for a seizure disorder with carbamazepine. Although his seizure frequency is decreased, valproic acid is added to further reduce the seizure frequency.
a. Is a drug–drug interaction of concern in this patient?
Patients are commonly treated with more than 1 drug, have individual dietary choices, and may also be using over-the-counter (OTC) medications, vitamins, and other “natural” supplements. This polypharmaceutical nature of health care requires consideration of potential drug interactions (see Figure 3-4).
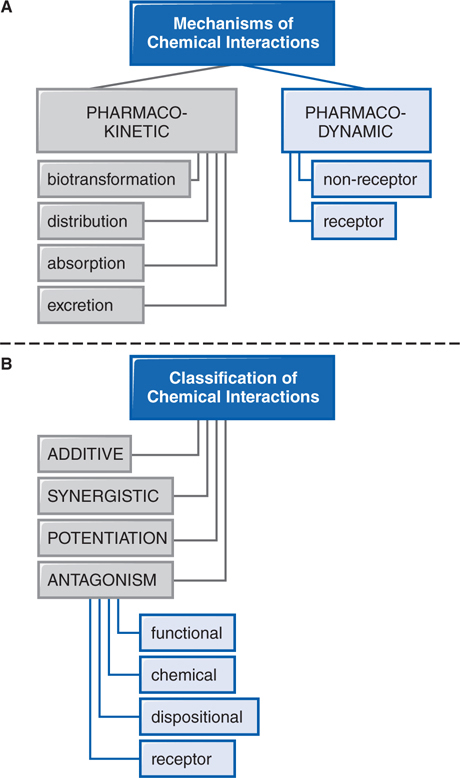
FIGURE 3-4 Mechanisms and classification of drug interactions.
A drug can frequently influence the metabolism of 1 or several other drugs (see Chapter 2), and this is especially notable with hepatic CYPs. Acetaminophen is partially transformed by CYP2E1 to the toxic metabolite NAPQI (see Figure 3-6). Intake of ethanol, a potent inducer of the 2E1 isoenzyme, may lead to increased susceptibility to acetaminophen poisoning after overdose. Similarly, a number of second-generation piperidine antihistamines (terfenadine, astemizole) were removed from the market when they were noted to lead to QT interval prolongation and tachydysrhythmias when coadministered with macrolide antibiotics.
b. What are the different types of drug–drug interactions?
Drug interactions generally involve drug absorption, protein binding, metabolism, receptor binding, or therapeutic action (see Side Bar DRUG–DRUG INTERACTIONS). Further drug–drug interactions can be classified as additive, synergistic, potentiation, or antagonism (see Side Bar CLASSIFICATION OF DRUG–DRUG INTERACTIONS).
CLASSIFICATION OF DRUG–DRUG INTERACTIONS
• Additive—the combined effect of 2 drugs equals the sum of the effect of each agent given alone
• Synergistic—the combined effect of 2 drugs exceeds the sum of the effects of each drug given alone
• Potentiation—the creation of a toxic effect of 1 drug due to the presence of another drug
• Antagonism—the interference of 1 drug with the action of another drug
▶ Chemical antagonism—reaction between 2 chemicals to neutralize their effects
▶ Dispositional antagonism—alteration of the disposition of a substance so that less of the agent reaches the target organ
▶ Receptor antagonism—blockade of the effect of 1 drug with another drug that competes at the receptor site
A 20-year-old man goes to a college party. A person at the party offers to spike his punch from an unlabeled flask. An hour later the man is experiencing an intense headache and blurred vision. He is taken to the emergency room where it is determined that he has been poisoned with methanol.
a. How does methanol produce toxicity?
One carbon alcohol, methanol (CH3OH), is also known as methyl and wood alcohol. It is an important industrial reagent and solvent found in products such as paint removers, shellac, and antifreeze; methanol is added to industrial-use ethanol to make it unsafe for human consumption.
Severe metabolic acidosis can develop due to the accumulation of formic acid (see Figure 3-5), and the respiratory depression can be severe, especially in the context of coma. The visual disturbances associated with methanol intoxication are a prominent part of the clinical picture and occur as a consequence of injury to ganglion cells of the retina caused by the metabolite, formic acid, with subsequent inflammation, atrophy, and potential bilateral blindness.
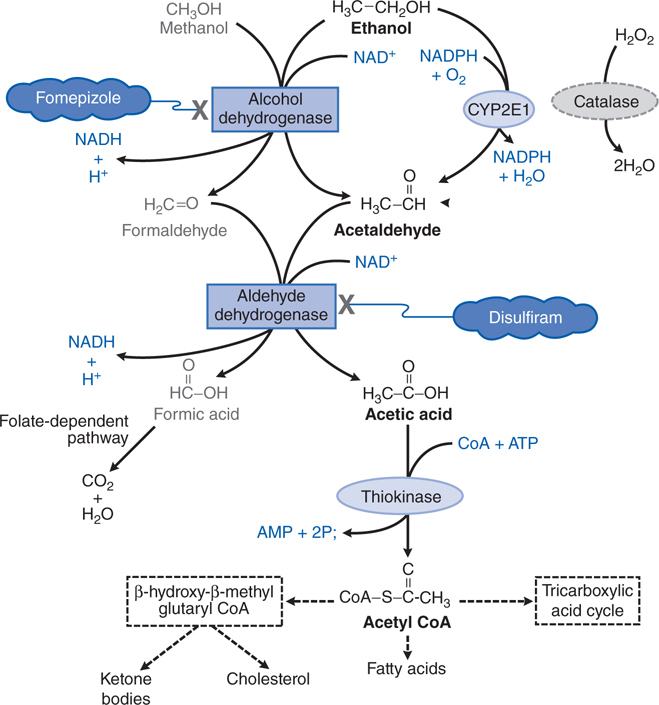
FIGURE 3-5 Metabolism of ethanol and methanol.
b. What are the signs and symptoms of methanol poisoning?
Methanol poisoning consists of headache, GI distress, and pain (partially related to pancreatic injury), difficulty breathing, restlessness, and blurred vision associated with hyperemic optic disks. The clinical picture can also include necrosis of the pancreas.
c. What are the treatment options for this patient?
Table 3-1 lists common antidotes and their indications. Methanol is rapidly absorbed via the oral route, inhalation, and through the skin, with the latter 2 routes being most relevant to industrial settings. Methanol also is metabolized (see Figure 3-5) by ADH and ALDH, with damaging consequences. Competition between methanol and ethanol for ADH forms the basis of the use of ethanol as an antidote in methanol poisoning. Fomepizole (4-methylpyrazole), an ADH inhibitor, is useful in methanol and ethylene glycol poisoning.
Following the inhibition of metabolism of methanol with either ethanol or fomepizole, hemodialysis is used to effectively remove the remaining methanol.
A 16-year-old woman is brought to the emergency room because she took an overdose of acetaminophen. Her vital signs are normal and she appears stable except for being distraught at having attempted suicide.
a. What are the general principles of the management of a poisoned patient?
The majority of poisoning exposures reported to US poison control centers are judged to be nontoxic or only minimally toxic. When toxicity is expected, or does occur, the priority of poisoning treatment is to support vital functions until the drug or chemical is eliminated from the body. Because of the acute onset of action and finite duration of action of most drugs, the treatment of poisoning must be prompt and goal directed. The first goal is to protect vital physiological functions from impairment. The second goal is to keep the concentration of poison in tissues as low as possible by preventing absorption and enhancing elimination. The third goal is to combat the toxicological effects of the poison at the effector sites.
Knowledge of the relative toxicity of drugs is the beginning to the proper management of a poisoned patient. The following tables put human poisonings in perspective: Table 3-4 lists the top 5 agents involved in drug-related deaths; Table 3-5 lists the substances most frequently involved in human poisoning exposures; Table 3-6 lists the poisons associated with the largest number of human fatalities.
TABLE 3-4 Top Five Agents Involved in Drug-Related Deaths
Cocaine
Opioids
Benzodiazepines
Alcohol
Antidepressants
U.S. DHHS.
TABLE 3-5 Substances Most Frequently Involved in Human Poisoning Exposures
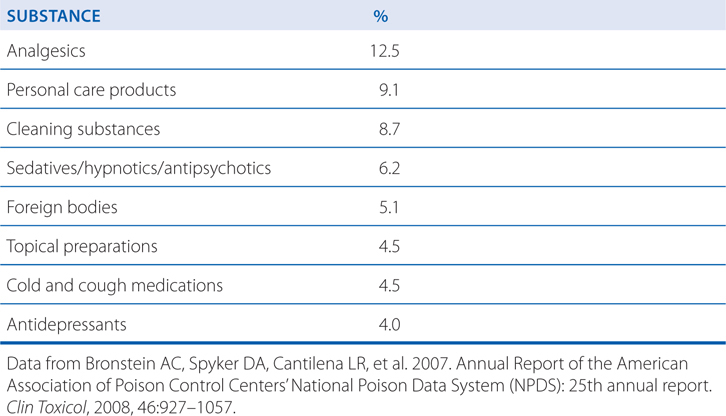
TABLE 3-6 Poisons Associated with the Largest Number of Human Fatalities
Sedatives/hypnotics/antipsychotics
Acetaminophen
Opioids
Antidepressants
Cardiovascular drugs
Stimulants and street drugs
Alcohols
Bronstein AC, Spyker DA, Cantilena LR, et al. 2007. Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 25th annual report. Clin Toxicol, 2008, 46:927–1057.
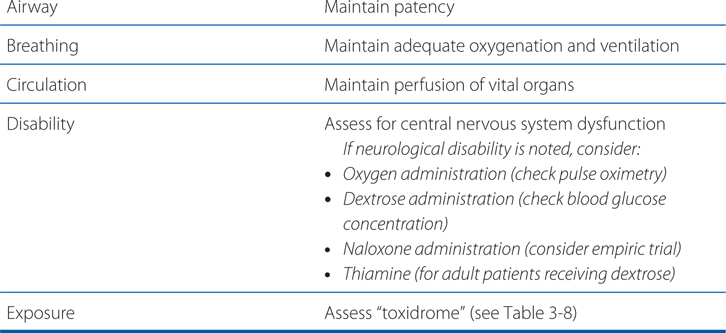
The “ABC” mnemonic of emergency care (airway, breathing, circulation) is popularly taught and applies to the treatment of acute poisoning, but with the addition of elements D (disability) and E (exposure) (see Table 3-7). In severe cases, endotracheal intubation, mechanical ventilation, pharmacological blood pressure support, and/or extracorporeal circulatory support may be necessary and appropriate.
TABLE 3-7 ABCDE: Initial Treatment Approach for Acute Poisoning
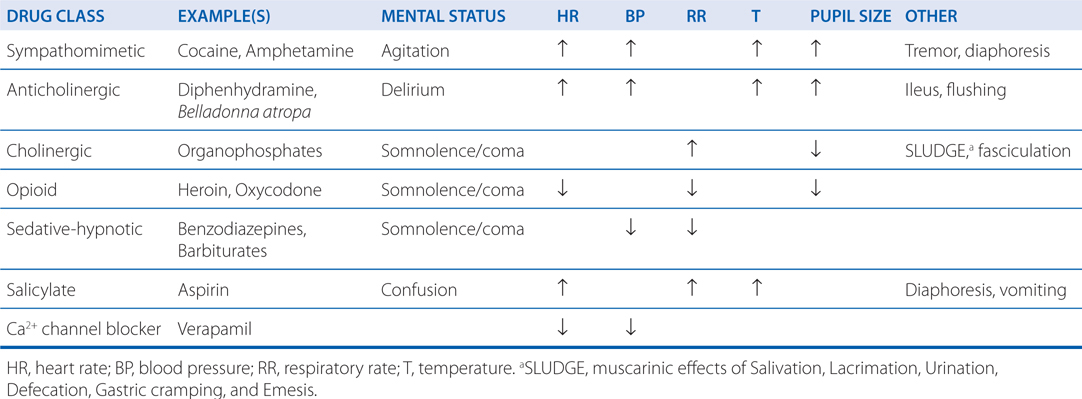
A carefully obtained medical history may create a list of available medications or chemicals that might be implicated in a poisoning event. Often, an observation of physical symptoms and signs may be the only additional clues to a poisoning diagnosis. Groups of physical signs and symptoms associated with specific poisoning syndromes are known as toxidromes. Table 3-8 describes commonly encountered toxidromes.
The most typically available urine drug toxicology test is an immunoassay designed to detect common drugs of abuse such as amphetamines, barbiturates, benzodiazepines, cannabis, cocaine, and opiates. Acute poisoning with these substances can usually be determined on clinical grounds, and the results of these assays are infrequently available fast enough to guide stabilization. Additionally, detection of drugs or their metabolites on a urine immunoassay does not mean that the detected drug is responsible for the currently observed poisoning illness. When ingestion of acetaminophen or aspirin cannot clearly be excluded via the exposure history, serum quantification of these drugs is recommended. An electrocardiogram (ECG) may be useful at detecting heart blocks, Na+ channel blockade, or K+ channel blockade associated with specific medication classes. Further laboratory analysis, such as use of blood gas determinations, serum chemistries, complete blood counts, and other testing, should be tailored to the individual poisoning circumstance.
b. How are the principles of absorption, distribution, metabolism, and elimination different for a drug after excessive exposure than after a therapeutic dose?
The principles of pharmacokinetics (absorption, distribution, metabolism, and elimination) are described in Chapter 2. Toxicokinetics (the pharmacokinetics of a drug under circumstances that produce toxicity or excessive exposure) may differ significantly after poisoning, and these differences may profoundly alter treatment decisions and prognosis. Ingesting larger than therapeutic doses of a pharmaceutical may prolong its absorption, alter its protein binding and apparent volume of distribution, and change its metabolic fate.
When confronted with a potential poisoning, 2 toxicokinetic questions should be foremost in the clinician’s mind:
• How long does an asymptomatic patient need to be monitored (drug absorption and dynamics)?
• How long will it take an intoxicated patient to get better (drug elimination and dynamics)?
c. How does acetaminophen produce toxicity?
Acetaminophen is metabolized to nontoxic glucuronide and sulfate conjugates, and to a highly reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI) via CYP isoforms. NAPQI is referred to as a biologically reactive intermediate, and such intermediates often arise due to drug metabolism. At therapeutic dosing, NAPQI binds to nucleophilic glutathione; but, in overdose, glutathione depletion may lead to the pathological finding of hepatic necrosis (see Figure 3-6). The highly reactive NAPQI metabolite binds covalently to cell macromolecules, leading to dysfunction of enzymatic systems and structural and metabolic disarray. Furthermore, depletion of intracellular GSH renders the hepatocytes highly susceptible to oxidative stress and apoptosis.
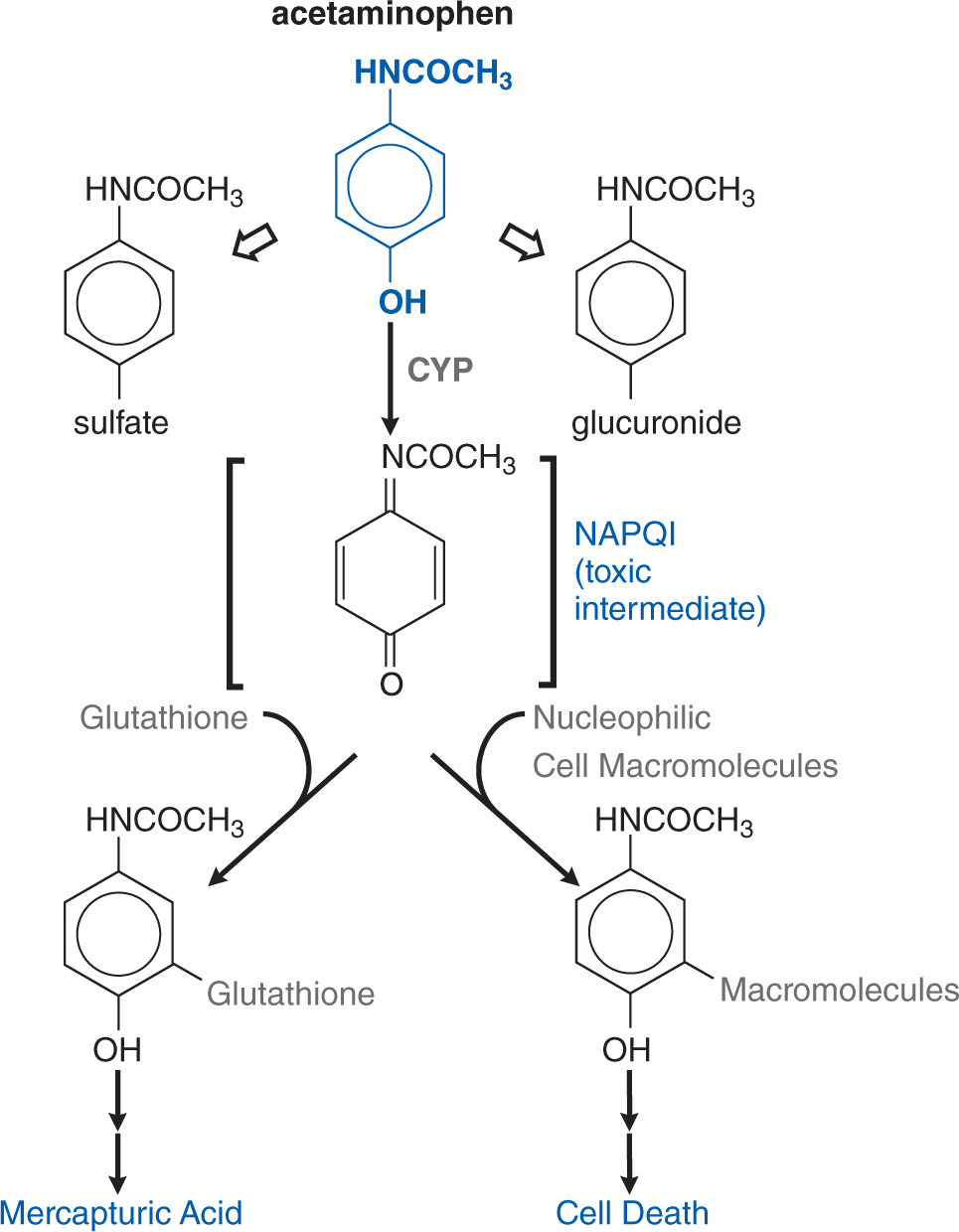
FIGURE 3-6 Pathways of acetaminophen metabolism and toxicity. The toxic intermediate NAPQI is N-acetyl-p-benzoquinoneimine.
Acute overdose can cause severe hepatic damage, and the number of accidental or deliberate poisonings with acetaminophen continues to grow. Chronic use of less than 2 g/day is not typically associated with hepatic dysfunction, but overuse of acetaminophen-containing narcotic and over-the-counter combination products marketed in the United States has led to heightened awareness of the possibility of toxicity.
Acute ingestion of more than 7.5 g of acetaminophen, or repeat use of supratherapeutic doses, can result in toxicity. The most serious acute adverse effect of an overdose of acetaminophen is a potentially fatal hepatic necrosis. Renal tubular necrosis and hypoglycemic coma also may occur.
In adults, hepatotoxicity may occur after ingestion of a single dose of 10 to 15 g (150-250 mg/kg) of acetaminophen; doses of 20 to 25 g or more are potentially fatal. Conditions of CYP induction (eg, heavy alcohol consumption) or GSH depletion (eg, fasting or malnutrition) increase the susceptibility to hepatic injury, which has been documented, albeit uncommonly, with doses in the therapeutic range.
Symptoms that occur during the first 2 days of acute poisoning by acetaminophen reflect gastric distress (eg, nausea, abdominal pain, anorexia) and belie the potential seriousness of the intoxication. Plasma hepatic transaminases become elevated, sometimes markedly so, beginning ~12 to 36 hours after ingestion. Clinical indications of hepatic damage manifest within 2 to 4 days of ingestion of toxic doses, with right subcostal pain, tender hepatomegaly, jaundice, and coagulopathy. Renal impairment or frank renal failure may occur. Liver enzyme abnormalities typically peak 72 to 96 hours after ingestion. The onset of hepatic encephalopathy or worsening coagulopathy beyond this time indicates a poor prognosis. Biopsy of the liver reveals centrilobular necrosis with sparing of the periportal area. In nonfatal cases, the hepatic lesions are reversible over a period of weeks or months.
d. How is the diagnosis of acetaminophen toxicity established?
Acetaminophen overdose constitutes a medical emergency. Severe liver damage occurs in 90% of patients with plasma concentrations of acetaminophen more than 300 μg/mL at 4 hours after ingestion or 45 μg/mL at 15 hours after the ingestion of the drug. Minimal hepatic damage can be anticipated when the drug concentration is less than 120 μg/mL at 4 hours or 30 μg/mL at 12 hours after ingestion.
e. How should acetaminophen poisoning be treated?
Early diagnosis and treatment of acetaminophen overdose is essential to optimize outcome. Perhaps 10% of poisoned patients who do not receive specific treatment develop severe liver damage; 10 to 20% of these eventually die of hepatic failure despite intensive supportive care. Activated charcoal, if given within 4 hours of ingestion, decreases acetaminophen absorption by 50 to 90% and is the preferred method of gastric decontamination. Gastric lavage generally is not recommended.
N-acetylcysteine (NAC)(see Table 3-1) is indicated for those at risk of hepatic injury. NAC therapy should be instituted in suspected cases of acetaminophen poisoning before blood concentrations become available, with treatment terminated if assay results subsequently indicate that the risk of hepatotoxicity is low. NAC functions by detoxifying NAPQI. It both repletes GSH stores and may conjugate directly with NAPQI by serving as a GSH substitute. There is some evidence that in cases of established acetaminophen toxicity, NAC may protect against extrahepatic injury by its antioxidant and anti-inflammatory properties. Even in the presence of activated charcoal, there is ample absorption of NAC, and neither should activated charcoal be avoided nor NAC administration be delayed because of concerns of a charcoal-NAC interaction.
Adverse reactions to NAC include rash (including urticaria, which does not require drug discontinuation), nausea, vomiting, diarrhea, and rare anaphylactic reactions. An oral loading dose of 140 mg/kg is given, followed by the administration of 70 mg/kg every 4 hours for 17 doses. Where available, the intravenous loading dose is 150 mg/kg by intravenous infusion in 200 mL of 5% dextrose over 60 minutes, followed by 50 mg/kg by intravenous infusion in 500 mL of 5% dextrose over 4 hours, then 100 mg/kg by intravenous infusion in 1000 mL of 5% dextrose over 16 hours.
In addition to NAC therapy, aggressive supportive care is warranted. This includes management of hepatic and renal failure if they occur and intubation if the patient becomes obtunded. Hypoglycemia can result from liver failure, and plasma glucose should be monitored closely. Fulminant hepatic failure is an indication for liver transplantation, and a liver transplant center should be contacted early in the course of treatment of patients who develop severe liver injury despite NAC therapy.
A 2-year-old-girl is brought to the emergency room after ingesting some of her pregnant mother’s iron tablets.
a. How should iron poisoning be established in this patient?
A history of ingestion by a parent is frequently sufficient to initiate therapy. A confirmation of the diagnosis is made with the measurement of a plasma iron.
In the evaluation of a child thought to have ingested iron, a color test for iron in the gastric contents and an emergency determination of the concentration of iron in plasma can be performed. If the latter is less than 63 μmol (3.5 mg/L), the child is not in immediate danger. However, vomiting should be induced when there is iron in the stomach, and an x-ray should be taken to evaluate the number of pills remaining in the small bowel (iron tablets are radiopaque).
Large amounts of ferrous salts are toxic, but fatalities are rare in adults. Most deaths occur in children, particularly between the ages of 12 and 24 months. As little as 1 to 2 g of iron may cause death, but 2 to 10 g usually is ingested in fatal cases. The frequency of iron poisoning relates to its availability in the household, particularly the supply that remains after a pregnancy. The colored sugar coating of many of the commercially available tablets gives them the appearance of candy. All iron preparations should be kept in childproof bottles.
b. What are the signs and symptoms of iron poisoning and how does iron produce toxicity?
Signs and symptoms of severe poisoning may occur within 30 minutes after ingestion or may be delayed for several hours. They include abdominal pain, diarrhea, or vomiting of brown or bloody stomach contents containing pills. Of particular concern are pallor or cyanosis, lassitude, drowsiness, hyperventilation due to acidosis, and cardiovascular collapse. If death does not occur within 6 hours, there may be a transient period of apparent recovery, followed by death in 12 to 24 hours. The corrosive injury to the stomach may result in pyloric stenosis or gastric scarring. Hemorrhagic gastroenteritis and hepatic damage are prominent findings at autopsy.
c. How should this patient be treated?
Iron in the upper GI tract can be precipitated by lavage with sodium bicarbonate or phosphate solution, although the clinical benefit is questionable. When the plasma concentration of iron is greater than the total iron-binding capacity (63 μmol; 3.5 mg/L), deferoxamine should be administered. Shock, dehydration, and acid-base abnormalities should be treated in the conventional manner. Most important is the speed of diagnosis and therapy. With early effective treatment, the mortality from iron poisoning can be reduced from as high as 45% to as low as 1%.
Deferoxamine is isolated as the iron chelate from Streptomyces pilosus and is treated chemically to obtain the metal-free ligand. Deferoxamine has the desirable properties of a remarkably high affinity for ferric iron (Ka = 1031) coupled with a very low affinity for calcium (Ka = 102). In vitro, it removes iron from hemosiderin and ferritin and, to a lesser extent, from transferrin. Iron in hemoglobin or cytochromes is not removed by deferoxamine.
Deferoxamine is poorly absorbed after oral administration, and parenteral administration is required. For severe iron toxicity (serum iron levels greater than 500 μg/dL), the intravenous route is preferred. The drug is administered at 10 to 15 mg/kg/hour by constant infusion. Faster rates of infusion (45 mg/kg/h) have been used in a few cases; rapid boluses usually are associated with hypotension. Deferoxamine may be given intramuscularly in moderately toxic cases (serum iron 350-500 μg/dL) at a dose of 50 mg/kg with a maximum dose of 1 g. Hypotension also can occur with the intramuscular route.
d. What are the toxic effects of deferoxamine therapy?
Deferoxamine causes a number of allergic reactions, including pruritus, wheals, rash, and anaphylaxis. Other adverse effects include dysuria, abdominal discomfort, diarrhea, fever, leg cramps, and tachycardia. Occasional cases of cataract formation have been reported. Deferoxamine may cause neurotoxicity during long-term, high-dose therapy for transfusion-dependent thalassemia major; both visual and auditory changes have been described. A “pulmonary syndrome” has been associated with high-dose (10-25 mg/kg/h) deferoxamine therapy; tachypnea, hypoxemia, fever, and eosinophilia are prominent symptoms. Contraindications to the use of deferoxamine include renal insufficiency and anuria; during pregnancy, the drug should be used only if clearly indicated.
A 48-year-old man has a community-acquired pneumonia. He is prescribed an oral penicillin product. Within 15 minutes of taking the first dose, he develops tightness in his chest, and numbness and swelling of his lips. His wife calls emergency services; he is treated in his home and transported to the hospital.
a. What are the different types of adverse reactions to medications?
The different types of toxicity to drugs include dose-dependent reactions that may be an extension of a drug’s therapeutic effect, pathological toxicity, genotoxicity, allergic reactions, and idiosyncratic reactions (see Side Bar TYPES OF THERAPEUTIC DRUG TOXICITY).
b. He does not remember ever-receiving penicillin before so what type of reaction is this?
He may have received penicillin as a child and not known what the medication was. An allergy is an adverse reaction that results from previous sensitization to a particular chemical or to one that is structurally similar. Such reactions are mediated by the immune system. For a low-molecular-weight chemical to cause an allergic reaction, it or its metabolic product usually acts as a hapten, combining with an endogenous protein to form an antigenic complex. Such antigens induce the synthesis of antibodies, usually after a latent period of at least 1 to 2 weeks. Subsequent exposure to the chemical results in an antigen-antibody interaction that provokes the typical manifestations of allergy. Dose-response relationships usually are not apparent for the provocation of allergic reactions. Allergic responses have been divided into 4 general categories based on the mechanism of immunological involvement (see Side Bar TYPES OF THERAPEUTIC DRUG TOXICITY).
Anaphylaxis is mediated by IgE antibodies. The Fc portion of IgE can bind to receptors on mast cells and basophils. If the Fab portion of the antibody molecule then binds antigen, various mediators (eg, histamine, leukotrienes, and prostaglandins) are released and cause vasodilation, edema, and an inflammatory response. The main targets of this type of reaction are the gastrointestinal (GI) tract (food allergies), the skin (urticaria and atopic dermatitis), the respiratory system (rhinitis and asthma), and the vasculature (anaphylactic shock). These responses tend to occur quickly after challenge with an antigen to which the individual has been sensitized and are termed immediate hypersensitivity reactions.
c. What would be the treatment for this type of reaction?
The use of epinephrine to immediately treat drug-induced anaphylaxis is introduced in Chapter 7 of Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition.
A 56-year-old woman previously developed a severe thrombocytopenia after receiving sulfamethoxazole-trimethoprim fixed-dose product (SEPTRA) for a urinary tract infection. She is now in the hospital for hip replacement surgery. Her hospital chart has a sticker “Allergic to SEPTRA” on the front cover. Following her surgery she develops a urinary tract infection and is given BACTRIM.
a. What is the issue with the administration of BACTRIM to this patient?
BACTRIM is another trade name for a fixed-dose combination of sulfamethoxazole-trimethoprim (see Chapter 38). So there should be concern that BACTRIM will cause the same type of adverse reaction as SEPTRA. This type of medication error is obviously preventable and stems from the lack of knowledge by hospital staff about the medications being administered. In this case, apparently the pharmacy staff either did not know that the patient had a history of a prior reaction to SEPTRA, or that BACTRIM was a similar product.
b. How can medication errors be avoided?
Over the past decade considerable attention has been given to the reduction of medication errors and adverse drug events (ADEs). Medication errors can occur in any part of the medication prescribing or use process, while ADEs are injuries related to the use or nonuse of medications. It is believed that medication errors are 50 to 100 times more common than ADEs. Some ADEs, such as previously unknown allergies, are unpreventable, but most medication errors can be prevented. Traditionally, the “5 Rights” of safe medication administration have been taught on hospital wards:
Right drug, right patient, right dose, right route, right time.
However, accomplishing a reduction in medication errors involves scrutiny of the systems involved in prescribing, documenting, transcribing, dispensing, administering, and monitoring a therapy.
c. How can medication errors be prevented?
Good medication use practices have mandatory and redundant checkpoints, such as having a pharmacist, a doctor, and a nurse all review and confirm that an ordered dose of a medication is appropriate for a patient prior to the drug’s administration. In such a system, medication errors occur only when several “holes” in the medication administration safeguards exist and are simultaneously aligned. Several practical strategies have been suggested to reduce medication errors within hospitals and other health care settings (see Table 3-9), and these strategies are being constantly revised.
TABLE 3-9 Best Practice Recommendations to Reduce Medication Administration Errorsa
Short Term
• Maintain unit-dose distribution systems for nonemergency medications
• Have pharmacies prepare intravenous solutions
• Remove inherently dangerous medications (eg, concentrated KCl) from patient care areas
• Develop special procedures for high-risk drugs
• Improve drug-related clinical information resources
• Improve medication administration education for clinicians
• Educate patients about the safe and accurate use of medications
• Improve access of bedside clinicians to pharmacists
Long Term
Implement technology-based safeguards:
• Computerized order entry
• Computerized dose and allergy checking
• Computerized medication tracking
• Use of bar codes or electronic readers for medication preparation and administration
aSee Massachusetts Hospital Association.
http://macoalition.org/documents/Best_Practice_Medication_Errors.pdf
HEALTH EFFECTS OF LEAD TOXICITY
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


