26 Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is a disease state characterised by airflow limitation that is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases (GOLD, 2009).
COPD has been defined (National Institute for Health and Clinical Excellence, 2010) as:
Epidemiology
COPD is the fifth leading cause of death in the UK and the fourth in the world. It is expected to rise to third position by 2020. It is estimated that over 3 million people have the disease in the UK, with 2 million having undiagnosed COPD (National Clinical Guideline Centre, 2010). COPD is the largest single cause of lost working days in the UK. It is accountable for more than 10% of all hospital admissions and directly costs the NHS around £491 m/year. The burden of COPD on the UK healthcare system exceeds that of asthma and is outlined in Table 26.1.
Table 26.1 Annual morbidity and mortality from COPD (National Clinical Guideline Centre, 2010)

Pathology
The major pathological changes in COPD affect four different compartments of the lung and all are affected in most individuals to varying degrees (American Thoracic Society/European Respiratory Society Task Force, 2004).
Aetiology
Not all smokers go on to develop clinically significant COPD; genetic factors seem to modify each individual’s risk. The age at which an individual begins smoking, total pack-years smoked and current smoking status are predictive of COPD mortality. Passive exposure to cigarette smoke may also contribute to respiratory symptoms and COPD by increasing the lungs’ total burden of inhaled particles and gases (GOLD, 2009). Tobacco exposure is quantified in ‘pack-years’:
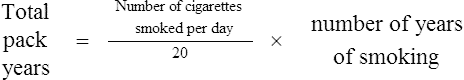
Additional risk factors include the natural ageing process of the lungs. Males are currently more at risk of developing chronic bronchitis, but as the number of women who smoke increases, the incidence of chronic bronchitis in females will also rise. The major risk factors are summarised in Table 26.2.
Table 26.2 Risk factors for the development of COPD
| Risk factor | Comment |
|---|---|
| Smoking | Risk increases with increasing consumption but there is also large interindividual variation in susceptibility |
| Age | Increasing age results in ventilatory impairment; most frequently related to cumulative smoking |
| Gender | Male gender was previously thought to be a risk factor but this may be due to a higher incidence of tobacco smoking in men. Women have greater airway reactivity and experience faster declines in FEV1, so may be at more risk than men |
| Occupation | The development of COPD has been implicated with occupations such as coal and gold mining, farming, grain handling and the cement and cotton industries |
| Genetic factors | α1-Antitrypsin deficiency is the strongest single genetic risk factor, accounting for 1–2% of COPD. Other genetic disorders involving tissue necrosis factor and epoxide hydrolase may also be risk factors |
| Air pollution | Death rates are higher in urban areas than in rural areas. Indoor air pollution from burning biomass fuel is also implicated as a risk factor, particularly in underdeveloped areas of the world |
| Socio-economic status | More common in individuals of low socio-economic status |
| Airway hyper-responsiveness and allergy | Smokers show increased levels of IgE, eosinophils and airway hyper-responsiveness but how these influence the development of COPD is unknown |
Pathophysiology
The pathogenic mechanisms and pathological changes described above lead to the physiological abnormalities of COPD: mucus hypersecretion, ciliary dysfunction, airflow limitation and hyperinflation, gas exchange abnormalities, pulmonary hypertension and systemic effects (American Thoracic Society/European Respiratory Society Task Force, 2004).
Airflow limitation and hyperinflation
This progressive destructive enlargement of the respiratory bronchioles, alveolar ducts and alveolar sacs is referred to as emphysema. Adjacent alveoli can become indistinguishable from each other, with two main consequences. The first is loss of available gas exchange surfaces, which leads to an increase in dead space and impaired gas exchange. The second consequence is the loss of elastic recoil in the small airways, vital for maintaining the force of expiration, which leads to a tendency for them to collapse, particularly during expiration. Increased thoracic gas volume and hyperinflation of the lungs result. The causes of airflow limitation in COPD are summarised in Box 26.1.
Clinical manifestations
Diagnosis
A diagnosis of COPD should be considered in any patient who has symptoms of cough, wheeze, regular sputum production or exertional dyspnoea and/or a history of exposure to COPD risk factors (see Table 26.2). Spirometry is then used to confirm the diagnosis. There is no single diagnostic test for COPD.
Investigations
Airflow obstruction is defined as:
Both UK and international COPD guidelines use spirometry to categorise the severity of COPD. These are summarised in Table 26.3. Testing should be carried out after a dose of inhaled bronchodilator to prevent overdiagnosis or overestimation of severity.
Table 26.3 Assessment of severity of airflow obstruction
| FEV1 | Severity (NICE) | Severity (GOLD) |
|---|---|---|
| Greater than 80% predicted | Stage I: Mild | |
| 50–80% predicted | Mild | Stage II: Moderate |
| 30–49% predicted | Moderate | Stage III: Severe |
| Less than 30% predicted | Severe | Stage IV: Very severe |
(adapted from National Institute for Health and Clinical Excellence, 2010; GOLD, 2009)
At diagnosis and evaluation, patients may receive other investigations as outlined in Table 26.4.
Table 26.4 Additional investigations at the diagnosis of COPD
| Investigation | Note |
|---|---|
| Chest X-ray | To exclude other pathologies |
| Full blood count | To identify anaemia or polycythaemia |
| Serial domiciliary peak flow measurements | To exclude asthma if there is a doubt about diagnosis |
| α1-Antitrypsin | Particularly with early-onset disease or a minimal smoking/family history |
| Transfer factor for carbon monoxide | To investigate symptoms that seem disproportionate to the spirometric impairment |
| CT scan of the thorax |



