21 Chronic heart failure
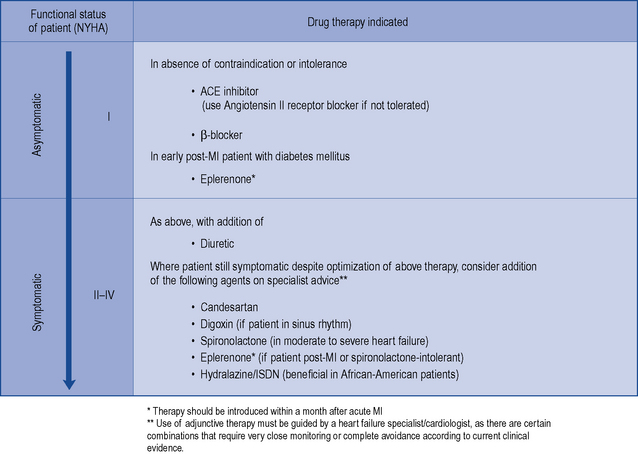
Treatment is aimed at improving left ventricular function, controlling the secondary effects that lead to the occurrence of symptoms, and delaying disease progression. Drug therapy is indicated in all patients with heart failure to control symptoms (where present), improve quality of life and prolong survival. Patients with heart failure usually have their functional status assessed and categorised using the New York Heart Association (NYHA) classification system shown in Table 21.1.
Table 21.1 New York Heart Association (NYHA) classification of functional status of the patient with heart failure
| I | No symptoms with ordinary physical activity (such as walking or climbing stairs) |
| II | Slight limitation with dyspnoea on moderate to severe exertion (climbing stairs or walking uphill) |
| III | Marked limitation of activity, less than ordinary activity causes dyspnoea (restricting walking distance and limiting climbing to one flight of stairs) |
| IV | Severe disability, dyspnoea at rest (unable to carry on physical activity without discomfort) |
Aetiology
Atrial fibrillation often accompanies hyperthyroidism and mitral valve disease, where a rapid and irregular ventricular response can compromise cardiac efficiency. Improved management of the underlying causes, where appropriate, may alleviate the symptoms of heart failure, whereas the presence of mechanical defects may require the surgical insertion of prosthetic valve(s). While around 50% of patients with heart failure have significant left ventricular systolic dysfunction, the other half is comprised of patients who have either a normal or insignificantly reduced left ventricular ejection fraction (EF), although there is no consensus on the threshold for compromised EF and assessment of each patient relies mainly on clinical symptoms. These patients are referred to as having heart failure with preserved left ventricular ejection fraction (HFPEF). However, most of the available evidence from clinical trials regarding the pharmacological treatment of heart failure to date relates to those patients with heart failure due to left ventricular systolic dysfunction. Clinical symptomatic description of chronic heart failure is mild, moderate, or severe heart failure. ‘Mild’ is used for patients who are mobile with no important limitations of dyspnoea or fatigue, ‘severe’ for patients who are markedly symptomatic in terms of exercise intolerance and ‘moderate’ for those with restrictions in between. Trials tend to formalise these categories into NYHA Categories I–IV (Table 21.1).
Clinical manifestations
Patients with heart failure may appear pale and their hands cold and sweaty. Reduced blood supply to the brain and kidney can cause confusion and contribute to renal failure, respectively. Hepatomegaly occurs from congestion of the gastro-intestinal tract, which is accompanied by abdominal distension, anorexia, nausea and abdominal pain. Oedema affects the lungs, ankles and abdomen. Signs of oedema in the lungs include crepitations heard at the lung bases. In acute heart failure, symptoms of pulmonary oedema are prominent and may be life-threatening. The sputum may be frothy and tinged red from the leakage of fluid and blood from the capillaries. Severe dyspnoea may be complicated by cyanosis and shock. Table 21.2 presents the clinical manifestations of heart failure.
Table 21.2 Clinical manifestations of heart failure
| Venous (congestion) | Cardiac (cardiomegaly) | Arterial (peripheral hypoperfusion) |
|---|---|---|
| Dyspnoea | Dilation | Fatigue |
| Oedema | Tachycardia | Pallor |
| Hypoxia | Regurgitation | Renal impairment |
| Hepatomegaly | Cardiomyopathy | Confusion |
| Raised venous pressure | Ischaemia, arrhythmia | Circulatory failure |
Investigations
Echocardiography is important when investigating patients with a suspected diagnosis of heart failure. An echocardiogram allows visualisation of the heart in real time and will identify whether heart failure is due to systolic dysfunction, diastolic dysfunction or heart valve defects. With the provision of direct access echocardiography services to doctors in primary care, an increasing number of patients can now be quickly referred to confirm or exclude heart failure due to left ventricular systolic dysfunction or other structural abnormalities. Some reports suggest that between 50% and 75% of patients referred to direct access clinics may have normal left ventricular function, which has important implications for the selection of appropriate drug treatment. Table 21.3 shows a number of investigations that are routinely performed in the assessment of heart failure symptoms. The use of serum natriuretic peptide measurement in the diagnosis of patients with heart failure is currently limited by the lack of defined cut-off values and, therefore, measurements are only considered in combination with ECG/chest X-ray data prior to echocardiography.
Table 21.3 Investigations performed to confirm a diagnosis of heart failure
| Investigation | Comment |
|---|---|
| Blood test | The following assessments are usually performed: |
| 12-lead electrocardiogram | A normal ECG usually excludes the presence of left ventricular systolic dysfunction. An abnormal ECG will require further investigation |
| Chest radiograph | A chest radiograph (X-ray) is performed to look for an enlarged cardiac shadow and consolidation in the lungs |
| Echocardiography | An echocardiogram is used to confirm the diagnosis of heart failure and any underlying causes, for example, valvular heart disease |
Treatment of heart failure
Until the 1980s, pharmacotherapy was driven by the aim to control symptoms, when diuretics and digoxin were the mainstay of treatment. While relieving the symptoms of heart failure remains decisive in improving a patient’s quality of life, a better understanding of the underlying pathophysiology has led to major advances in the pharmacological treatment of heart failure. With the introduction of angiotensin converting enzyme (ACE) inhibitors, β-blockers, angiotensin II receptor blockers (ARBs) and aldosterone antagonists, delaying disease progression and ultimately improving survival have become realistic goals of therapy. An outline of the site of action of the various drugs is schematically presented in Fig. 21.1.
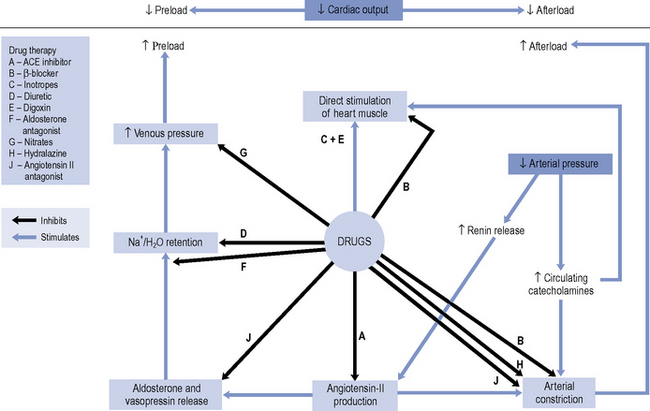
There is consensus that all patients with left ventricular systolic dysfunction should be treated with both an ACE inhibitor and a β-blocker in the absence of intolerance or contraindications. The evidence base for treatment clearly shows that use of an ACE inhibitor (or angiotensin receptor blocker) and β-blocker therapy in patients with heart failure due to left ventricular systolic dysfunction leads to an improvement in symptoms and reduction in mortality. There is some evidence to suggest that either agent can be initiated first, as both appear to be just as effective and well tolerated (CARMEN 2004, CIBIS III, 2005). Beneficial effects on morbidity and mortality have also been shown for the use of ARBs, aldosterone antagonists and hydralazine/nitrate combinations when used in the treatment of chronic heart failure. Digoxin has been shown to improve morbidity and reduce the number of hospital admissions in patients with heart failure, although its effect on mortality has not been demonstrated. Table 21.4 describes the treatment of acute heart failure in the hospital setting, while Fig. 21.2 highlights the possible treatment options for patients with chronic heart failure due to left ventricular systolic dysfunction.
Table 21.4 Treatment of acute heart failure due to left ventricular systolic dysfunction in patients requiring hospitalisation
| Problem | Drug therapy indicated |
|---|---|
| Anxiety | Use of intravenous opiates to reduce anxiety and reduce preload through venodilation |
| Breathlessness | High-flow oxygen (60–100%) may be required in conjunction with i.v. furosemide as either direct injection or 24-h infusion (5–10 mg/h). |
| Venodilation with i.v. GTN is also effective at doses titrated every 10–20 min against systolic BP ≤ 110 mmHg | |
| Arrhythmia | Digoxin useful in control of atrial fibrillation. Amiodarone is the drug of choice in ventricular arrhythmias |
| Expansion of blood volume following blood transfusion | An elevation in preload, such as can occur acutely by expansion of blood volume after a transfusion, can exacerbate the degree of systolic dysfunction. Therefore, it is necessary to continue or increase diuretic dosage during this time |
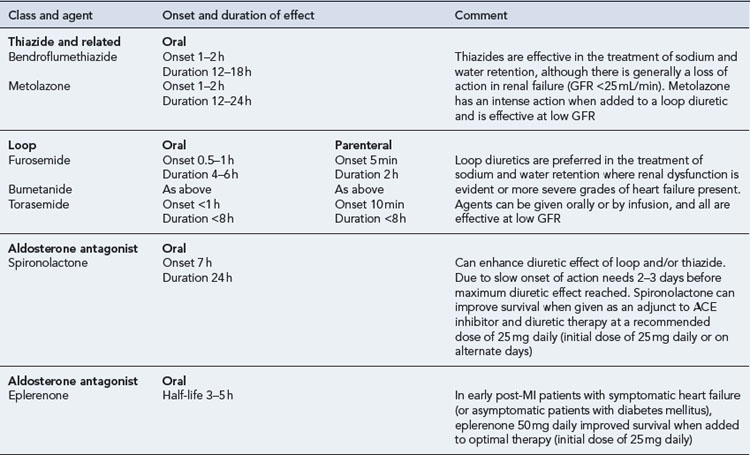
Diuretics
Details of diuretic therapy used in left ventricular systolic dysfunction are summarised in Table 21.5.
ACE inhibitors
ACE inhibitors are generally well tolerated by most patients and have been shown to improve the quality of life and survival in patients with mild-to-severe systolic dysfunction (CONSENSUS, 1987; CONSENSUS II, 1992; SOLVD-P, 1992; SOLVD-T, 1991; V-HeFT II, 1991), including those patients who have experienced a myocardial infarction (AIRE), 1993; SAVE, 1992; TRACE, 1995). When an ACE inhibitor is prescribed, it is important to ensure that the dose is started low and increased gradually, paying close attention to renal function and electrolyte balance. The dose should be titrated to achieve the target dose that has been associated with long-term benefits shown in clinical trials or (if not possible) the maximum tolerable dose. There is some evidence to suggest that high doses of ACE inhibitor are more effective than low doses in relation to reduction in mortality, although it is uncertain whether this is a general class effect (ATLAS, 1999). In clinical practice, it is possible that some patients may be treated with ACE inhibitors at doses below those used in clinical trials. As a consequence, actual outcomes in heart failure treatment may not be as good as expected from the trial findings.
The introduction of an ACE inhibitor may produce hypotension, which is most pronounced after the first dose and is sometimes severe. Patients at risk include those already on high doses of loop diuretics, where the diuretics cannot be stopped or reduced beforehand, and patients who may have a low-circulating fluid volume (due to dehydration) and an activated renin–angiotensin system. Hypotension can also occur where the ACE inhibitor has been initiated at too high a dose or where the dose has been increased too quickly after initiation. In the primary care setting, treatment must be started with a low dose which is usually administered at bedtime. In patients at particular risk of hypotension, a test dose of the shorter-acting agent captopril can be given to assess suitability for treatment before commencing long-term treatment with a preferred ACE inhibitor. Once it has been established that the ACE inhibitor can be initiated safely, the preferred option would be to switch to a longer acting agent with once- or twice-daily dosing, starting with a low dose which would be gradually titrated upwards to the recommended target (Table 21.6). Monitoring of fluid balance, blood biochemistry and blood pressure are essential safety checks during initiation and titration of ACE inhibitor therapy.
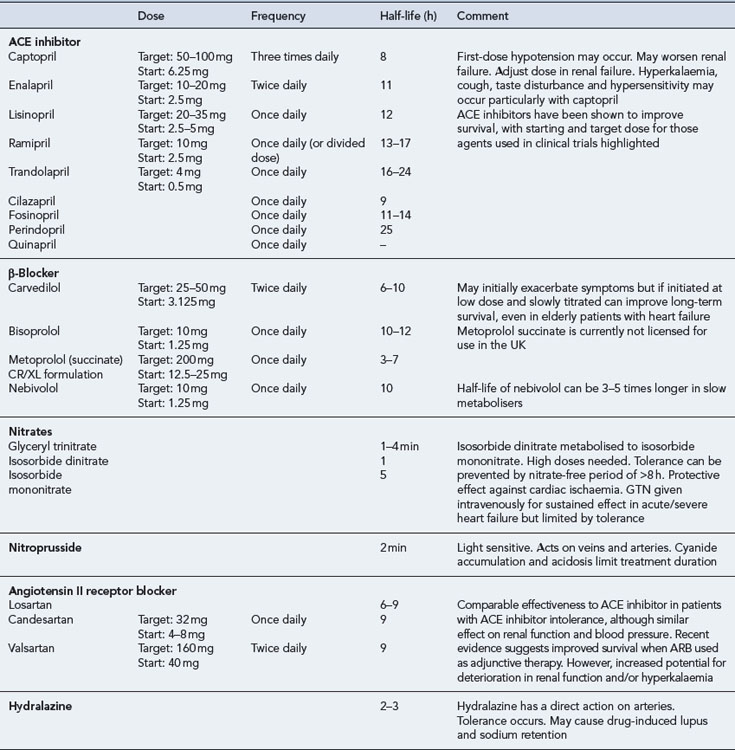
ACE inhibitors are potentially hazardous in patients with pre-existing renal disease, as blockade of the renin-angiotensin system may lead to reversible deterioration of renal function. In particular, ACE inhibitors are contraindicated in patients with bilateral renal artery stenosis, in whom the renin-angiotensin system is highly activated to maintain renal perfusion. Since most ACE inhibitors or their active metabolites rely on elimination via the kidney, the risk of other forms of dose-related toxicity is also increased in the presence of renal failure. Fosinopril, which is partially excreted by metabolism, may be the preferred agent in patients with renal failure. ACE inhibitors are also contraindicated in patients with severe aortic stenosis because their use can result in a markedly reduced cardiac output due to decreased filling pressure within the left ventricle. Table 21.6 summarises the activity and use of ACE inhibitors.
Angiotensin II receptor blockers
The use of ARBs as an adjunct to ACE inhibitor and β-blocker therapy has been associated with significant reductions in cardiovascular events and hospitalisation rate (CHARM Added, 2003). Although this finding is encouraging, the impact on mortality alone remains inconsistent and there is no clear consensus on when to use an ARB as adjunctive therapy. In studies involving patients unable to tolerate an ACE inhibitor, ARBs have been shown to be comparable to ACE inhibitors in reducing the risk of cardiovascular death and rate of hospitalisation, and in the control of symptoms in heart failure patients (CHARM Alternative, 2003; Val-HeFT, 2002). Therefore, ARBs are recommended for use as an alternative to ACE inhibitor therapy where intolerance has been confirmed. It is important to note that in patients who have renal failure secondary to ACE inhibitors, switching to an ARB is of no theoretical or practical benefit, as similar adverse effects are likely.
A recent meta-analysis has raised concerns about a possible increase of cancer in people taking ARBs (Sipahi et al., 2010). Although the implications of this are unclear it adds weight to the recommendation that ACE inhibitors, not ARBs, should be the first-line agent when selecting a drug to act on the renin-angiotensin system.
β-Blockers
Formerly, β-blockers have been contraindicated in patients with heart failure. However, the sympathetic neurohormonal overactivity that occurs in response to the failing heart has been identified as a decisive factor in the progression of ventricular dysfunction. Consequently, β-blockers have been tested in a number of clinical trials. There is now substantial evidence that β-blockers reduce mortality among patients with mild-to-moderate symptomatic heart failure (ANZ Carvedilol, 1997; CAPRICORN, 2001; CIBIS II, 1999; MERIT-HF, 1999; US Carvedilol, 1996) and those with severe heart failure (COPERNICUS, 2001). This beneficial effect also extends to the elderly heart failure population (SENIORS, 2005).
The use of β-blockers is, therefore, recommended for all patients with heart failure due to left ventricular systolic dysfunction, irrespective of age and the degree of dysfunction. However, due to their negative inotropic effects, β-blockers should only be initiated when the patient’s condition is stable. There is insufficient evidence for a class effect to be assumed illustrated by the fact that in one trial, metoprolol tartrate was found to be inferior to carvedilol (COMET, 2003). Currently, nebivolol, bisoprolol and carvedilol are the only licensed β-blockers for the treatment of heart failure in the UK.
It is likely that patients will experience a worsening of symptoms during initiation of therapy and, therefore, patients are started on very low doses of β-blocker (e.g. carvedilol 3.125 mg daily) with careful titration occurring over a number of weeks or months with careful monitoring. The goal is to titrate the dose towards those used in clinical trials that have been associated with morbidity and mortality benefits (carvedilol 25–50 mg daily). Table 21.6 summarises the activity and use of β-blockers in heart failure.
Despite the demonstrated benefits, there is ongoing concern that certain subgroups of patients with heart failure continue to be undertreated with β-blockers. These groups include patients with chronic obstructive pulmonary disease (COPD), peripheral vascular disease, diabetes mellitus, erectile dysfunction and older adults. With the exception of patients with reversible pulmonary disease, who have typically been excluded from β-blocker trials (CIBIS II, 1999; MERIT-HF, 1999), there is now sufficient evidence to justify the use of β-blockers licensed for heart failure in these patients. In addition, a systematic review of trials on cardio-selective β-blockers found no clinically significant adverse respiratory effects in patients with reversible COPD, although it would be prudent to use these agents in such patients with caution and with appropriate monitoring in place (Salpeter S. et al., 2005).
Aldosterone antagonists
The use of aldosterone antagonists as an adjunct to standard treatment has been shown to have an effect on morbidity and mortality in patients with heart failure. Spironolactone has been shown to reduce mortality and hospitalisation rates in patients with moderate-to-severe heart failure (RALES, 1999). The use of eplerenone has also been shown to be associated with similar benefits in early post-MI patients with symptomatic heart failure or early post-MI diabetic patients with asymptomatic heart failure (EPHESUS, 2003, EMPHASIS-HF, 2010).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


