Chapter 6 Choosing the target
Introduction: the scope for new drug targets
How many drug targets are there?
A compendium of therapeutic drugs licensed in the UK and/or US (Dollery, 1999) lists a total of 857 compounds (Figure 6.1), of which 626 are directed at human targets. Of the remainder, 142 are drugs used to treat infectious diseases, and are directed mainly at targets expressed by the infecting organism, and 89 are miscellaneous agents such as vitamins, oxygen, inorganic salts, plasma substitutes, etc., which are not target-directed in the conventional sense.
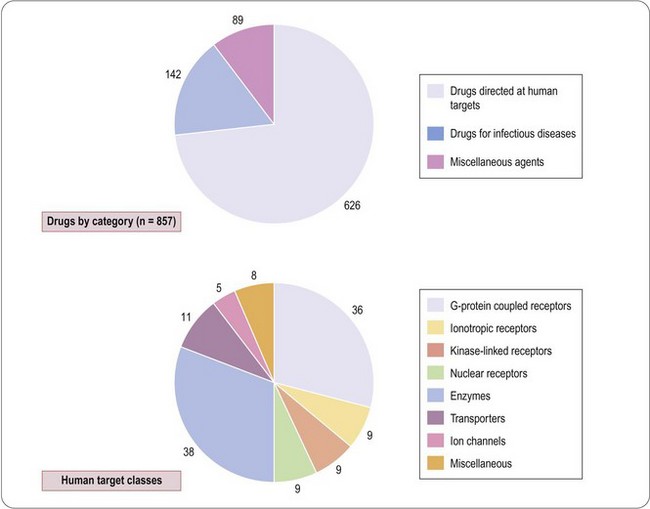
Drews and Ryser (1997) estimated that these drugs addressed approximately 500 distinct human targets, but this figure includes all of the molecular subtypes of the generic target (e.g. 14 serotonin receptor subtypes and seven opioid receptor subtypes, most of which are not known to be therapeutically relevant, or to be specifically targeted by existing drugs), as well as other putative targets, such as calcineurin, whose therapeutic relevance is unclear. Drews and Ryser estimated that the number of potential targets might be as high as 5000–10 000. Adopting a similar but more restrictive approach, Hopkins and Groom (2002) found that 120 drug targets accounted for the activities of compounds used therapeutically, and estimated that 600–1500 ‘druggable’ targets exist in the human genome. From an analysis of all prescribed drugs produced by the 10 largest pharmaceutical companies, Zambrowicz and Sands (2003) identified fewer than 100 targets; when this analysis was restricted to the 100 best-selling drugs on the market, the number of targets was only 43, reflecting the fact that more than half of the successful targets (i.e. those leading to therapeutically effective and safe drugs) failed to achieve a significant commercial impact. More recently, Imming et al. (2006) identified 218 targets of registered drugs, including those expressed by infectious agents, whereas Overington et al. (2006) found 324 (the difference depending on the exact definition of what constitutes a single identified target). How many targets remain is highly uncertain and Zambrowicz and Sands (2003) suggested that during the 1990s only two or three new targets were exploited by the 30 or so new drugs registered each year, most of which addressed targets that were already well known. The small target base of existing drugs, and the low rate of emergence of new targets, suggests that the number yet to be discovered may be considerably smaller than some optimistic forecasters have predicted. They estimated that 100–150 high-quality targets in the human genome might remain to be discovered. However, a recent analysis of 216 therapeutic drugs registered during the decade 2000–2009 (Rang unpublished) showed that 62 (29%) were first in class compounds directed at novel human targets. This is clearly an increase compared with the previous decade and may give grounds for optimism that many more new targets remain to be found. In the earlier analysis by Drews and Ryser (1997), about one-quarter of the targets identified were associated with drugs used to treat infectious diseases, and belonged to the parasite, rather than the human, genome. Their number is even harder to estimate. Most anti-infective drugs have come from natural products, reflecting the fact that organisms in the natural world have faced strong evolutionary pressure, operating over millions of years, to develop protection against parasites. It is likely, therefore, that the ‘druggable genome’ of parasitic microorganisms has already been heavily trawled – more so than that of humans, in whom many of the diseases of current concern are too recent for evolutionary counter-measures to have developed.
More recent estimates of the number of possible drug targets (Overington et al., 2006; Imming et al., 2006) are in line with the figure of 600–1500 suggested by Hopkins and Groom (2002). The increased use of gene deletion mutant mice and si RNA for gene knock down coupled with a bioinformatic approach to protein characterization is starting to increase the reliability of target identification (Imming et al., 2006; Wishart et al., 2008; Bakheet and Doig, 2009). New approaches, in which networks of genes are associated with particular disease states, are likely to throw up additional targets that may not have been discovered otherwise (e.g. see Emilsson et al., 2008).
The nature of existing drug targets
The human targets of the 626 drugs, where they are known, are summarized in Figure 6.1. Of the 125 known targets, the largest groups are enzymes and G-protein-coupled receptors, each accounting for about 30%, the remainder being transporters, other receptor classes, and ion channels. This analysis gives only a crude idea of present-day therapeutics, and underestimates the number of molecular targets currently addressed, since it fails to take into account the many subtypes of these targets that have been identified by molecular cloning. In most cases we do not know which particular subtypes are responsible for the therapeutic effect, and the number of distinct targets will certainly increase as these details become known. Also there are many drugs whose targets we do not yet know. There are a growing number of examples where drug targets consist, not of a single protein, but of an oligomeric assembly of different proteins. This is well established for ion channels, most of which are hetero-oligomers, and recent studies show that G-protein-coupled receptors (GPCRs) may form functional dimers (Bouvier, 2001) or may associate with accessory proteins known as RAMPs (McLatchie et al., 1998; Morphis et al., 2003), which strongly influence their pharmacological characteristics. The human genome is thought to contain about 1000 genes in the GPCR family, of which about one-third could be odorant receptors. Excluding the latter, and without taking into account the possible diversity factors mentioned, the 40 or so GPCR targets for existing drugs represent only about 7–10% of the total. Roughly one-third of cloned GPCRs are classed as ‘orphan’ receptors, for which no endogenous ligand has yet been identified, and these could certainly emerge as attractive drug targets when more is known about their physiological role. Broadly similar conclusions apply to other major classes of drug target, such as nuclear receptors, ion channels and kinases.
The above discussion relates to drug targets expressed by human cells, and similar arguments apply to those of infective organisms. The therapy of infectious diseases, ranging from viruses to multicellular parasites, is one of medicine’s greatest challenges. Current antibacterial drugs – the largest class – originate mainly from natural products (with a few, e.g. sulfonamides and oxazolidinediones, coming from synthetic compounds) first identified through screening on bacterial cultures, and analysis of their biochemical mechanism and site of action came later. In many cases this knowledge remains incomplete, and the molecular targets are still unclear. Since the pioneering work of Hitchings and Elion (see Chapter 1), the strategy of target-directed drug discovery has rarely been applied in this field1; instead, the ‘antibiotic’ approach, originating with the discovery of penicillin, has held sway. For the 142 current anti-infective drugs (Figure 6.1), which include antiviral and antiparasitic as well as antibacterial drugs, we can identify approximately 40 targets (mainly enzymes and structural proteins) at the biochemical level, only about half of which have been cloned. The urgent need for drugs that act on new targets arises because of the major problem of drug resistance, with which the traditional chemistry-led strategies are failing to keep up. Consequently, as with other therapeutic classes, genomic technologies are being increasingly applied to the problem of finding new antimicrobial drug targets (Rosamond and Allsop, 2000; Buysse, 2001). In some ways the problem appears simpler than finding human drug targets. Microbial genomes are being sequenced at a high rate, and identifying prokaryotic genes that are essential for survival, replication or pathogenicity is generally easier than identifying genes that are involved in specific regulatory mechanisms in eukaryotes.
Conventional strategies for finding new drug targets
Two main routes have been followed so far:
There are numerous examples where the elucidation of pathophysiological pathways has pointed to the existence of novel targets that have subsequently resulted in successful drugs, and this strategy is still adopted by most pharmaceutical companies. To many scientists it seems the safe and logical way to proceed – first understand the pathway leading from the primary disturbance to the appearance of the disease phenotype, then identify particular biochemical steps amenable to therapeutic intervention, then select key molecules as targets. The pioneers in developing this approach were undoubtedly Hitchings and Elion (see Chapter 1), who unravelled the steps in purine and pyrimidine biosynthesis and selected the enzyme dihydrofolate reductase as a suitable target. This biochemical approach led to a remarkable series of therapeutic breakthroughs in antibacterial, anticancer and immunosuppressant drugs. The work of Black and his colleagues, based on mediators and receptors, also described in Chapter 1, was another early and highly successful example of this approach, and there have been many others (Table 6.1a). In some cases the target has emerged from pharmacological rather than pathophysiological studies. The 5HT3 receptor was identified from pharmacological studies and chosen as a potential drug target for the development of antagonists, but its role in pathophysiology was at the time far from clear. Eventually animal and clinical studies revealed the antiemetic effect of drugs such as ondansetron, and such drugs were developed mainly to control the nausea and vomiting associated with cancer chemotherapy. Similarly, the GABAB receptor (target for relaxant drugs such as baclofen) was discovered by analysis of the pharmacological effects of GABA, and only later exploited therapeutically to treat muscle spasm.
Table 6.1 Examples of drug targets identified, (a) by analysis of pathophysiology, and (b) by analysis of existing drugs
| (a) Targets identified via pathophysiology | ||
| Disease indication | Target identified | Drugs developed |
| AIDS | Reverse transcriptase HIV protease | Zidovudine Saquinavir |
| Asthma | Cysteinyl leukotriene receptor | Zafirlukast |
| Bacterial infections | Dihydrofolate reductase | Trimethoprim |
| Malignant disease | Dihydrofolate reductase | 6-mercaptopurine Methotrexate |
| Depression | 5HT transporter | Fluoxetine |
| Hypertension | Angiotensin-converting enzyme Type 5 phosphodiesterase Angiotensin-2 receptor | Captopril Sildenafil Losartan |
| Inflammatory disease | COX-2 | Celecoxib Rofecoxib |
| Alzheimer’s disease | Acetylcholinesterase | Donepezil |
| Breast cancer | Oestrogen receptor | Tamoxifen Herceptin |
| Chronic myeloid leukemia | Abl kinase | Imatinib |
| Parkinson’s disease | Dopamine synthesis MAO-B | Levodopa Selegiline |
| Depression | MAO-A | Moclobemide |
| (b) Targets identified via drug effects | ||
| Drug | Disease | Target |
| Benzodiazepines | Anxiety, sleep disorders | BDZ binding site on GABAA receptor |
| Aspirin-like drugs | Inflammation, pain | COX enzymes |
| Ciclosporin, FK506 | Transplant rejection | Immunophilins |
| Vinca alkaloids | Cancers | Tubulin |
| Dihydropyridines | Cardiovascular disease | L-type calcium channels |
| Sulfonylureas | Diabetes | KATP channels |
| Classic antipsychotic drugs | Schizophrenia | Dopamine D2 receptor |
| Tricyclic antidepressants | Depression | Monoamine transporters |
| Fibrates | Raised blood cholesterol | PPARα |
The identification of drug targets by the ‘backwards’ approach – involving analysis of the mechanism of action of empirically discovered therapeutic agents – has produced some major breakthroughs in the past (see examples in Table 6.1b). Its relevance is likely to decline as drug discovery becomes more target focused, though natural product pharmacology will probably continue to reveal novel drug targets.
It is worth reminding ourselves that there remain several important drug classes whose mechanism of action we still do not fully understand (e.g. acetaminophen (paracetamol)2, valproate). Whether their targets remain elusive because their effects depend on a cocktail of interactions at several different sites, or whether novel targets will emerge for such drugs, remains uncertain.
New strategies for identifying drug targets
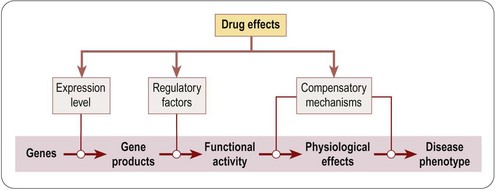
Figure 6.2 summarizes the main points at which drugs may intervene along the pathway from genotype to phenotype, namely by altering gene expression, by altering the functional activity of gene products, or by activating compensatory mechanisms. This is of course an oversimplification, as changes in gene expression or the activation of compensatory mechanisms are themselves indirect effects, following pathways similar to that represented by the primary track in Figure 6.2. Nevertheless, it provides a useful framework for discussing some of the newer genomics-based approaches. A useful account of the various genetic models that have been developed in different organisms for the identification of new drug targets, and elucidating the mechanisms of action of existing drugs, is given by Carroll and Fitzgerald (2003).
Trawling the genome
• Splice variants may result in more than one pharmacologically distinct type of receptor being encoded in a single gene. These are generally predictable from the genome, and represented as distinct species in the transcriptome and proteome.
• There are many examples of multimeric receptors, made up of non-identical subunits encoded by different genes. This is true of the majority of ligand-gated ion channels, whose pharmacological characteristics depend critically on the subunit composition (Hille, 2001). Recent work also shows that G-protein-coupled receptors often exist as heteromeric dimers, with pharmacological properties distinct from those of the individual units (Bouvier, 2001). Moreover, association between receptors and non-receptor proteins can determine the pharmacological characteristics of certain G-protein-coupled receptors (McLatchie et al., 1998).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


