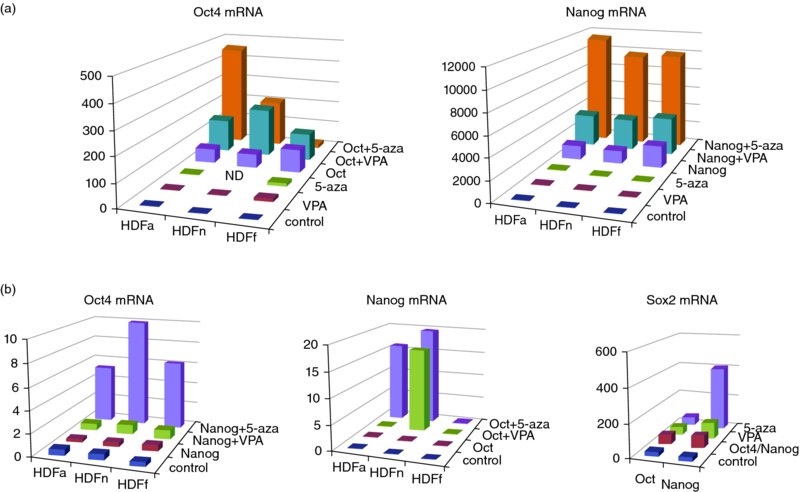
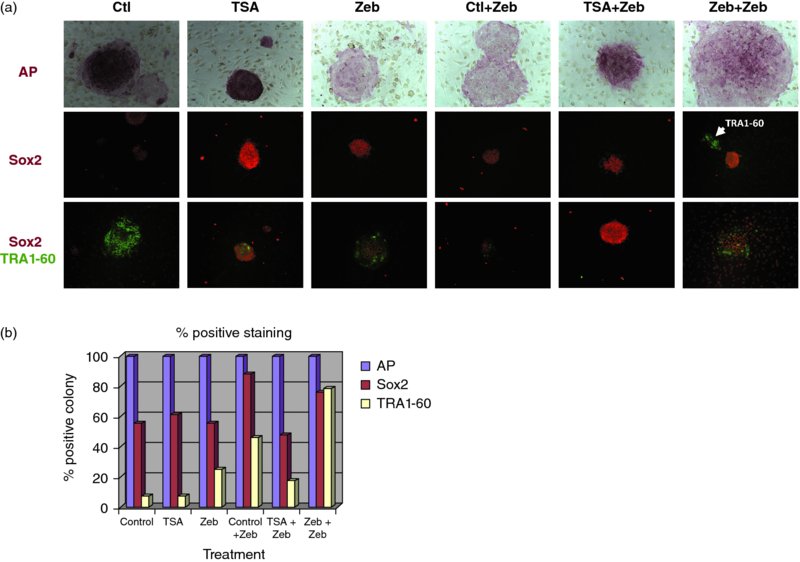
10 Jeong-Do Kim1, Jong S. Rim1, Robert B. Crochet2, Yong-Hwan Lee2, Jaroslaw Staszkiewicz1, Ru Gao1, and Kenneth J. Eilertsen1,3 1NuPotential, Inc., USA 2Department of Biological Science, Louisiana State University, USA 3 Pennington Biomedical Research Center, LSU System, USA A variety of reprogramming strategies are being developed that address the safety concerns associated with protocols dependent on genetic alterations such as virus-based reprogramming. Approaches include the use of nonintegrating plasmids and mRNA [1–6], microRNA [7], excisable transposons [8,9], and proteins [10,11]. An alternative method to enhance reprogramming is the use of a small-molecule inhibitor, or a cocktail of small-molecule inhibitors. Several chemicals have been reported to either improve reprogramming efficiency or substitute for specific factors. DNA and histone methyltransferase inhibitors, in particular, have shown tremendous promise in improving reprogramming efficiencies and generating safer induced pluripotent stem cells (iPSCs). These enzymes play important roles in controlling gene expression and are gaining attention in drug discovery and medicinal chemistry due to their association with a variety of pathologies. The objective of our studies is to identify novel lead compounds for human DNMT1 and histone methyltransferase inhibition targeting the S-adenosylmethionine (SAM) binding pocket of these enzymes, utilizing computer-based approaches. Reprogramming of mouse and human fibroblasts is an inefficient process. For mouse embryonic fibroblasts (MEFs), Mikkelson and colleagues reported that only a small percentage of cells transduced with defined transcription factors reactivated endogenous Nanog. Moreover, the majority of infected cells appeared to be trapped in a partially reprogrammed state [12–14]. They further demonstrated that partially reprogrammed cells could be induced to become fully reprogrammed by treatment with the DNA methyltransferase inhibitor 5-aza-cytidine (5-AZA). In addition, treatment with 5-AZA improved embryonic stem cell (ESC)-like colony formation by fourfold. We have proposed that epigenetic modification increases somatic cell reprogramming efficiency through (i) direct activation of pluripotent gene expression and (ii) activation of a pluripotent transcription factor network that is unique in pluripotent stem cells, including ESCs [15]. To test this hypothesis, we used a combination of two epigenetic modifying agents: a DNMT inhibitor and a histone-modifying enzyme inhibitor [15]. Primary cultures from three different origins of human dermal fibroblast (adult, neonatal, and fetal) were treated with epigenetic modifying agents with and without ectopic expression of Oct4 and Nanog genes. As shown in Figure 10.1, inhibition of DNMT in human dermal fibroblast cultures induced the expression of Oct4, Nanog, and Sox2 through auto- (Figure 10.1a) and reciprocal activation of the endogenous pluripotent transcription factor network (Figure 10.1b). In addition, DNMT inhibitor treatment, both before and after lentiviral-induced pluripotent factor expression, increased the efficiency of colony maturation by measuring co-expression of Sox2 (an early pluripotent marker) and TRA1-60 (a late pluripotent marker) from alkaline phosphatase (AP)-positive colonies (Figure 10.2). These results indicate that DNA methylation is an important epigenetic barrier that can limit the production of fully reprogrammed cells and that reprogramming efficiency can be improved by inhibiting specific repressive epigenetic regulatory components at the level of ESC-like colony formation and maturation. DNA methylation in mammalian cells occurs through the transfer of a methyl group from the universal cofactor SAM to cytosine of CpG dinucleotides [16,17] and is catalyzed by two types of DNA methyltransferase: a maintenance methyltransferase, DNMT1, and de novo methyltransferases, DNMT3a and 3b [18,19]. Figure 10.1 Real-time RT-PCR analysis. (a) Oct4 (left) and Nanog (right) expression in human dermal fibroblasts. Primary cultures from human dermal fibroblasts (HDFa, adult; HDFn, neonatal; HDFf, fetal) that transfected with or without Oct4 lentivirus (right) or Nanog lentivirus (right) were treated with VPA or 5-azacytidine as indicated. Total RNA was isolated and subjected to quantitative RT-PCR. Expression levels were compared to known standard samples and normalized by GAPDH. (b) Oct4 (left), Nanog (middle), and Sox2 (right) expression in human dermal fibroblasts. Primary human dermal fibroblasts that transfected with or without Nanog lentivirus (left) or Oct4 lentivirus (middle) were treated with VPA or 5-azacytidine as indicated. For Sox2 expression (right), HDFf was transfected with lentivirus for Oct4 or Nanog overexpression, then treated with VPA or 5-azacytidine as indicated. Total RNA was isolated and subjected to quantitative RT-PCR. Expression levels were compared to known standard samples and normalized by GAPDH Figure 10.2 Generation and characterization of AdiPSCs from human preadipocytes. (a) Morphology of ESC-like colonies with AP staining, single (Sox2) or double (Sox2, TRA1-60) immunostaining from TSA, and Zebularine pre- and post-treatment. (b) Percentage of positive colonies from AP staining and Sox2 and TRA1-60 immunostaining DNMT inhibitors are categorized into two groups: (i) nucleoside analogs, which incorporate into DNA and/or RNA and inhibit DNA methylation by trapping DNMTs, and (ii) nonnucleoside analogs, which directly inhibit DNMT activity by blocking the SAM pocket of the enzyme [20,21]. Only two DNMT inhibitors derived from the nucleoside cytidine, 5-azacytidine, and 5-aza-2′-deoxycytidine (decitabine) are currently in clinical trials for anticancer therapy [22,23]. However, despite their use in the successful treatment of cancers such as acute lymphoblastic leukemia and myelodysplastic syndrome, these drugs have shown severe cytotoxicity in patients due to random incorporation into all deoxycytidine and cytidine sites of DNA and/or RNA, followed by DNMT depletion [24–26]. Most DNMT inhibitors were identified by serendipity. In contrast, RG108 was the first DNMT inhibitor identified and developed by virtual screening using 3D homology modeling of the catalytic domain of DNMT1. RG108 has shown comparatively low cytotoxicity in human cell lines, but the inhibition rate of RG108 for the purified DNMT1 is low (<20%) compared to that of S-adenosylhomocysteine (SAH) (> 70%), a demethylated form of SAM [27,28]. There is a need to develop next-generation DNMT inhibitors for therapeutic and reprogramming applications. Our efforts to develop a new class of DNMT inhibitor are centered on the use of computer-based approaches with an experimental crystal structure (3SWR). Protein lysine methylation is a key signaling mechanism in eukaryotic cells. Protein lysine methyltransferases (PKMTs) catalyze the transfer of a methyl group from SAM to the ϵ-amino group of lysine residues of proteins, including histones [29]. PKMTs show substantial variations in protein substrate selectivity and the degree of methylation on lysine from mono- to di- to trimethylation. At present few small-molecule probes exist to dissect the functionality of PKMTs. Histone methyltransferases can be subdivided into three classes: the SET domain-containing lysine methyltransferases and the non-SET-domain lysine methyltransferases and arginine methyltransferases (PRMTs). The focus of this chapter is on histone lysine methyltransferases. All of them use SAM as a cosubstrate for the methylation reaction and generate SAH as a byproduct. Histone methylation does not alter the charge of the lysine residue in question. However, it does affect the basicity, hydrophobicity, and size of the amino acid side-chain group, which affects the affinity of proteins that recognize such side chains [30]. Although these changes are subtle, there are proteins that are able to bind selectively to certain methylated residues based on specific structural motifs [31] (for review see [32]). Chromodomains recognize mono-, di-, or trimethylated lysine residues; Tudor domains bind trimethylated lysines; plant homeo domain (PHD) fingers bind di- or trimethylated lysines; malignant brain tumor (MBT) domains bind mono- and dimethylated lysines. Histone lysine methylation is associated with both transcriptional activation and repression. For example, methylation of the tumor-suppressor protein p53 on K370 by Smyd2 [33] or on K382 by SET8 [34] leads to inhibition of transcriptional activity, whereas methylation on K372 by Set9 results in transcriptional activation [35]. The lysine methyltransferase G9a has recently been shown to dimethylate p53 on K373. This was correlated with inactivity of p53 [36]. Its impact on apoptotic processes and its overexpression in various cancer types suggest that G9a is a putative oncogene. G9a is a Histone3 Lysine9 (H3K9) methyltransferase and has been reported to be overexpressed in a variety of cancers [36–38]. G9a has also been shown to be involved in cocaine addiction [39], mental retardation [40], and DNA methylation in mouse ESCs. GLP is a closely related protein which shares 80% sequence identity with G9a and forms a heterodimer with it. Both G9a and GLP catalyze mono- and dimethylation of H3K9. Inhibition of G9a and GLP using small-molecule inhibitors has been demonstrated to facilitate the reprogramming of mouse neural precursor cells (NPCs) into iPSCs. Specfically, the G9a methyltransferase inhibitor BIX-01294 (BIX) was shown to improve the reprogramming efficiencies of OK (Oct4, Klf4)-infected NPCs by nearly eightfold [41]. G9a has been reported to mediate the repression of Oct4 [42] and Nanog [43]. Thus, it is possible that G9a inhibition could facilitate the reactivation of silenced Oct4 and Nanog and promote reprogramming. The beneficial effect of BIX on reprogramming was further substantiated using MEFs [44]. BIX treatment in combination with the DNA methyltransferase inhibitor RG108 and L-calcium channel agonist BayK8644 (BayK) signficantly enhanced the reprogramming of BIX-treated MEFs transduced with Oct4 and Klf4. Thus, the combination of epigenetic inhibitors with other target inhibitors may be required to overcome resistance to reprogramming and induce responsiveness. The development of selective, potent, and cell-active chemical inhibitors would be extremely valuable in furthering the efficiency and elucidating the mechanisms of somatic cell reprogramming. In addition, suppression of the histone methyltransferases DOT1L and SUV39H1 improved reprogramming. In particular, inhibition of the H3K79 histone methyltransferase DOT1L by shRNA or a small-molecule inhibitor increased the number of iPSC colonies, while substituting for KLF4 and MYC [45]. Onder et al. further reported that fibroblast-specific genes associated with the epithelial–mesenchymal transition lost H3K79me2 and that DOT1L inhibition facilitated the loss of the mark from genes typically repressed in the pluripotent state. These studies further suggest that specific chromatin-modifying enzymes can be modulated to improve the efficiency of reprogramming and that the production of iPSCs using only small molecules may be possible. Methyltransferases can be inhibited by analogs of the methyl group donor SAM. There are three known cosubstrate analogs that inhibit a variety of methyltransferases: (i) sinefungin, an antibiotic compound that is structurally similar to SAM; (ii) SAH, the demethylated cosubstrate of SAM, which serves as a feedback inhibitor; and (iii) methylthioadenosine. All of these inhibitors lack selectivity [46]. There are only a few known drug-like inhibitors of lysine methyltransferases, most of which were discovered by random screening approaches. The first inhibitor was chaetocin, a fungal mycotoxin. It inhibits both the Drosophila melanogaster enzyme Su(var)3–9 with an IC50 of 0.8 mM and G9a with an IC50 of 2.5 mM [47]. The compound BIX-01294 was identified in a combined virtual and high-throughput screening approach and shown to be an inhibitor of G9a with an IC50 of 3 mM. Cultured cells treated with BIX-01294 showed a reduction of histone H3K9 dimethylation and a decrease in cell number. In the same screening procedure, the compound BIX-01338 was discovered as a nonselective inhibitor of methyltransferases. It did not show any selectivity between lysine and arginine methyltransferases with an IC50 of 5 mM for G9a [48].
Chemicals Facilitating Reprogramming: Targeting the SAM Binding Site to Identify Novel Methyltransferase Inhibitors
10.1 Introduction
10.2 DNA Methyltransferases, Inhibition, and Reprogramming
10.3 DNMT Inhibitors
10.4 Histone Methyltransferases, Inhibition, and Reprogramming
10.5 Inhibitors of Lysine Methyltransferases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


