Overview
Enzymes are specialized proteins, which accelerate or catalyze a biochemical reaction. Each enzyme catalyzes a specific reaction and is regulated by competitive and noncompetitive inhibitors and/or by allosteric molecules. Multiple enzymes can catalyze a series of consecutive reactions, known as pathways, to produce and/or break down complex biological molecules. Examples include amino acid synthesis and degradation, the coordinated reactions involved in protein synthesis, and the urea cycle. Problems with enzyme pathways can not only lead to disease but also offer the opportunity for disease treatment via medications, which target specific points in these pathways.
Enzymes
Enzymes bring together one or more molecules, called substrates, to form a resulting molecule called a product (Figure 5-1). Most enzymes catalyze one specific reaction. However, some multipart enzyme complexes catalyze a series of step-by-step reactions—the first enzyme passes its product, now a new substrate, to a second enzyme that is part of the complex, the second passes its product to a third and so on. Enzymes are responsible for many essential reactions in the human body; in fact, there are from 20,000 to 25,000 total human genes, with about 25% of them producing enzymes. It is not surprising that problems with enzymes are caused by or result in diseases.
Figure 5-1.
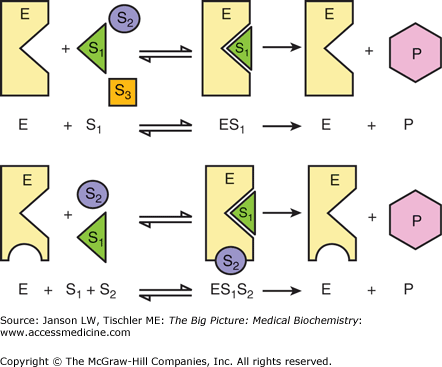
Illustration of Simple One and Two Substrate Enzyme Reaction. Components include enzyme (E), substances (S1, S2, S3), and product (P). If the substance can bind to the enzyme’s substrate-binding site (e.g., S1, top figure, and S1 and S2, bottom figure), then it acts as a substrate. Molecular shape as determined by secondary, tertiary, and quaternary structure as well as the hydrophobic/hydrophilic and neutral or charged nature influences which substrate molecules can bind as substrates (represented graphically by differing shapes of S1, S2, and S3 and the triangular and circular binding pockets). The enzyme binds the substrate molecule(s), catalyzes the enzymatic reaction, and releases the product after which the enzyme is ready to catalyze the same reaction again. The rate of the overall reaction is influenced by each step in the process, including substrate binding, rate of the reaction, and product release as discussed in the text below. [Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
The concept of enzyme kinetics allows an exact description of the enzymatic reaction, including the influence of substrate and product molecules and how fast the enzyme catalyzes the reaction and the impact. More advanced enzyme kinetics allows the mathematical expression of how other molecules such as cofactors, inhibitors, and activators (see below) affect the enzyme reaction.
For example, as discussed in Chapter 1, specific amino acids form an enzyme’s primary structure and, therefore, secondary to quaternary structure. These structures, in turn, form substrate-binding sites (also known as the “active site”) to accommodate the substrates, their chemical reaction, and the departure of the product. Substrates and products can both be a hydrophobic, hydrophilic, charged, uncharged, or neutral molecule, or a combination of the above. Mutations that result in the change of an amino acid or amino acids that make the substrate pocket can drastically change the enzyme’s activity. For example, a hydrophobic substrate will easily enter an enzyme pocket that is lined with hydrophobic amino acids. However, if one of these hydrophobic amino acids is changed to a highly charged amino acid, the substrate may not be able to enter the pocket and the enzyme may no longer function. How well the substrate interacts with the substrate-binding site represents the “affinity” of the enzyme for the substrate. The stronger the affinity, the less substrate is needed to achieve a certain rate of reaction. This concept is important if a mutation changes the substrate-binding site and lowers the affinity. This concept is illustrated in Figure 5-1.
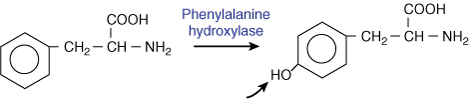
Substrate Concentration and Enzyme-based Diseases: Phenylketonuria (PKU) is an important autosomal recessive disease occurring in an average of 1 in 15,000 births. This genetic disease is usually caused by the deficiency of the enzyme phenylalanine hydroxylase, required to convert phenylalanine to tyrosine (see reaction below; hydroxyl group indicated by small arrowhead), important in the production of neurotransmitters and skin pigment (melanin). However, the disease does not result only from the decrease of tyrosine levels; rather, the resulting high concentration of phenylalanine leads to the activity of an otherwise minor enzymatic pathway, which produces phenylketones, including phenylacetate, phenylpyruvate, and phenylethylamine. The presence of these phenylketones is normally screened for in the blood as part of standard neonatal testing and, again, at 2 weeks of age.
[Adapted with permission from Kibble JD and Halsey CR: The Big Picture: Medical Physiology, 1st edition, McGraw-Hill, 2009.] Patients with PKU usually express traits of albinism, i.e., very fair skin; white-blond hair, and notable, pale blue eyes; a “musty” odor from phenylacetate in their sweat, urine, skin, and hair; and sometimes develop eczema. Untreated PKU results in decreased brain development (microcephaly), hyperactivity, brain damage, seizures, and severe learning disabilities/mental retardation. PKU is usually treated with a diet low in phenylalanine content, although some feel that residual phenylalanine levels may still cause neurological damage. As a result, additional treatments are being developed, which further decrease phenylalanine levels. |
The number of molecules of substrate increases or decreases the likelihood of the substrate interacting with the enzyme and, therefore, the rate of the reaction. When all of the enzyme molecules have substrate occupying the binding sites, the enzyme is said to be saturated by substrate and the rate of the reaction is maximized. If lower amounts of substrate are present, the reaction would also be slower than normal, potentially leading to clinical manifestations. If a disease state causes a molecule to abnormally increase in concentration, it may become a substrate for an enzyme with which it would normally not react.
The final factor that decides maximum rate of the reaction is the efficiency at which an enzyme can catalyze a single reaction. Enzyme mutations can diminish this efficiency, although, in rare instances, a mutation can increase activity. Likewise, the cell has regulatory molecules that can change the rate of the reaction by decreasing or increasing the efficiency of the enzyme.
Enzyme reactions often require additional molecules called cofactors (Figure 5-2). Examples of these cofactors include vitamins, nicotinamide adenine dinucleotide (NADH), nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FADH2), and coenzyme A (CoA). For example, during an enzyme reaction, NADH or NADPH donates a hydrogen atom as well as electrons to the product and becomes NAD+ or NADP+, respectively, and are then released. FADH2 also donates electrons and two hydrogen atoms to produce FAD. Conversely, NAD+, NADP+, and FAD accept electrons to become the respective reduced forms. Some inorganic metal ions such as iron (Fe), copper (Cu), calcium (Ca), manganese (Mn), magnesium (Mg), or zinc (Zn) also serve as cofactors for reactions. Metal ions may contribute or accept an electron (e.g., Fe2+ → Fe3+) or their associated charge may simply provide essential stabilization for the chemical reaction without changing the metal.
Figure 5-2.
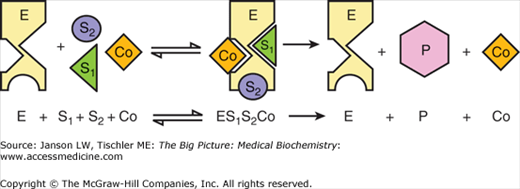
Enzymatic Reaction Involving a Cofactor. Components include enzyme (E), substrates (S1, S2), cofactor (Co), and product (P). The cofactor is necessary for the creation of the final product but is unchanged at the end of the reaction. [Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
Diseases of Copper Deficiency: The importance of proper levels of copper (Cu2+) to the human body is often not appreciated. For example, the incorporation of copper into several enzymes in mitochondria is essential for oxidative phosphorylation (Chapter 6), and low levels can adversely affect the production of ATP. The opposite, an excess of copper, leads to the important clinical conditions of Wilson’s disease and Menke’s “kinky hair” disease. Wilson’s disease results from low activity of ATPase, Cu2+ transporting, beta polypeptide (ATP7B), which is responsible for copper transport out of cells. The transported copper is carried by ceruloplasmin in the circulation. In Wilson’s disease, copper ion concentrations are increased in liver, brain, eyes, and other tissues. Liver deposits result in fatigue, confusion from high ammonia levels (hepatic encephalopathy), heightened blood pressure (portal hypertension), an increased risk of bleeding from blood vessels in the esophagus (esophageal varices), and, eventually, liver failure and/or cancer. Accumulation in the brain can lead to deterioration of memory and thought processes, loss of muscle control and tone, seizures, migraines, depression, anxiety, psychosis, and, often, the tremors and slow movement of Parkinson’s disease. Deposition in eyes results in the diagnostic “Kayser–Fleischer ring,” a brownish-green ring evident in the cornea. Kidneys, heart, and parathyroid glands can also be adversely affected by copper deposits. Menke’s “kinky hair” disease is a very rare, X-linked recessive disorder that mutates ATPase, Cu2+ transporting, alpha polypeptide (ATP7A) Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|