Fig. 15.1
General hallmarks of aging. A detailed explanation of the relevance of this hallmarks and its particularization to ARHL is given in the text (Reproduced with permission of Cell)
It is beyond the scope of this chapter to discuss in detail the multiplicity of factors contributing to aging at the cellular level, their combinations and interactions, which have become known from the use of model organisms from yeasts and worms to primates (for excellent recent reviews, see Gems and Partridge 2013; López-Otín et al. 2013). In Fig. 15.1, an outline of these factors in relation to ARHL is provided in order to place the factors that may contribute to ARHL within the much larger and complex, multifactorial nature of general aging. The relative contributions of each of these factors and potential therapeutic implications for prevention of ARHL have not yet been explored in adequate detail and represent targets for future research.
Factors contributing to aging are interconnected in complex networks. However, identification of hierarchical levels may facilitate operative categorizations. At the top of the proposed “hierarchical network” are the cumulative, combined effects of genome damage and associated genomic instability, altered epigenetic regulation, and defective protein homeostasis (for detailed discussion of the genetics of ARHL, see Chap. 14 by RD Frisina and DR Frisina). An interesting proposal is that these genetic factors may be the main “primers” of the aging process as a whole (López-Otín et al. 2013) (Fig. 15.1).
Accumulation of lesions to the genome, both nuclear and mitochondrial, due to sustained exogenous and endogenous damage and mutations, leading to imbalance of DNA repair and/or maintenance mechanisms, and associated unstable DNA transcription and genomic regulation when these mechanisms fail (Fig. 15.1), is central to age-related cell death (López-Otín et al. 2013). Disruption of nuclear architecture due to mutations in nuclear lamin genes contributes to age-associated genome instability, and mitochondrial DNA damage and mutations are of central relevance in ARHL.
A related primary cause of cellular damage associated with aging is impaired epigenetic regulation (López-Otín et al. 2013) (Fig. 15.1), likely due to damage to the enzymatic systems involved. Altered DNA methylation, aberrant posttranslational modifications of histones, or chromatin remodeling, all may add to genomic instability through faulty transcription, aberrant RNA processing, or defective DNA repair, with profound impact in key cell regulatory pathways (López-Otín et al. 2013). Understanding of epigenetic regulatory mechanisms and their impairment with age in the auditory system is still in its infancy (Provenzano and Domann 2007; Friedman and Avraham 2009; Rossman and Klein 2011), although the possible relevance of alterations in the epigenetic modification of histones during auditory aging has been recently demonstrated with the finding of a switch from acetylated to dimethylated histone H3 in the cochlea of aged mice (Watanabe and Bloch 2013).
A third major, primary factor associated with aging is impaired protein homeostasis (Fig. 15.1) (López-Otín et al. 2013). Sustained endogenous and exogenous cellular stress alters normal protein folding, overriding the molecular chaperones necessary for quality control and repair to assure proper folding and ubiquitination, proteasomal degradation, and regulated autophagy necessary for the disposal of misfolded proteins. Impaired refolding of damaged proteins or altered degradation mechanisms may lead to accumulation of abnormal proteins and subsequent cellular damage. In this regard, the heat shock transcription factor HSF1, the key regulator of the stress response mediated by heat shock proteins (Hsp) involved in protein folding repair, is intensely expressed in the cochlea (Fairfield et al. 2002). There is evidence that HSF1 and Hsps regulated by this transcription factor, notably those acting as molecular chaperones in the regulation of protein polymerization and folding, like Hsp27, Hsp70, or Hsp90, are involved in the stress response associated with different insults to the cochlea (Altschuler et al. 2002; Sugahara et al. 2003; Fairfield et al. 2004, 2005; Gong et al. 2012). The precise role of these mechanisms in cell death and loss of function (i.e., ARHL) remains elusive however. Pharmacological induction of Hsp expression attenuates age-related progression of hearing loss in some mice strains, albeit in a limited fashion (Mikuriya et al. 2008). Consistent with this, dysregulated autophagic stress with diminished autophagy activity and impaired removal of misfolded, aberrant proteins may also participate in age-related cell damage in spiral ganglion neurons (SGN) (Menardo et al. 2012).
A second set of factors involved in age-related damage likely is limited or exhausted cellular homeostatic protective mechanisms active against cumulative, sustained damage (Fig. 15.1). Such loss of protective mechanisms results in dysregulation of pathways for sensing and adapting metabolism to ongoing cell needs, mitochondrial dysfunction, and altered regulation of cellular senescence (Fig. 15.1) (López-Otín et al. 2013), which are relevant for ARHL as well.
Several signaling pathways involved in the control of cell growth and metabolic and energetic state, aimed at adapting cell needs to changing environmental conditions, show age-dependent dysregulations. Insulin-like growth factor-1 (IGF-1) is a major regulator of cell growth and differentiation, mediating the effects of growth hormone. IGF-1 shares signaling pathways with insulin. Risking oversimplification, insulin/IGF-1 signaling may be seen as an “anabolic sensor” system that couples rates of cell metabolism and growth to nutrient (glucose) abundance. It has been proposed that under conditions favoring damage like those operating during aging, the observed downregulation of insulin/IGF-1 signaling with age may be an adaptation to diminish metabolism and cell growth, thus limiting the cellular consequences of such damage (Schumacher et al. 2008; Garinis et al. 2008; López-Otín et al. 2013) and allowing lifespan extension. Below a given limit, however, such downregulation no longer provides a defensive role in aging but becomes incompatible with cell homeostasis and life. This is an attractive hypothesis that needs further experimental investigation. The impact of IGF-1 signaling in ARHL is complex, but IGF-1 appears to be protective in both mice and humans (for reviews see Bao and Ohlemiller 2010; Varela-Nieto et al. 2013). Studies in IGF-1 knockout mice show that IGF-1 is needed for the maintenance of hearing and that age-related decreasing levels of this growth factor parallel the progression of ARHL (Riquelme et al. 2010).
Another “anabolic sensor” mechanism relevant to aging includes the mTOR (mammalian target of rapamycin) intracellular cascade. mTOR kinases are themselves involved in the cascade of IGF-1 intracellular signaling. However, one of its functional forms, mTORC1, also participates in adapting cell growth and metabolism to amino acid concentrations through a complex pathway involving the lysosomal membrane (Efeyan et al. 2012). Genetic manipulations or pharmacological inhibition of mTOR with rapamycin extends lifespan in many animal models (Harrison et al. 2009). Increased mTOR activity contributes to age-related obesity (Yang and Ming 2012). Therefore, it seems that intense trophic and anabolic activity signaled through the insulin/IGF-1 or mTORC1 pathways are major contributors to aging, although their specific roles in ARHL remain to be examined in detail.
Two other metabolic state-sensing pathways acting in opposition to the insulin/IGF-1 and mTOR pathways are also relevant to aging. A pathway involving AMPK (5′AMP-dependent protein kinase) signals low-energy availability in cells by detecting high AMP levels. Pathways involving sirtuins (see below) also signal low energy by detecting high NAD+ levels (Houtkooper et al. 2010, 2012). Therefore, both AMPK and sirtuins detect limited nutrient availability and they signal catabolism predominance in cells. Their upregulation extends lifespan and seems to favor healthy aging in several aging models (López-Otín et al. 2013). Actually, the long known benefits of dietary or caloric restriction (CR) in extending life- and health span in many aging organisms (for a recent review, see Szafranski and Mekhail 2014) seem to be linked to the upregulation of AMPK and sirtuins and downregulation of mTOR and the insulin/IGF-1 pathway, triggered by limitation of nutrient availability, with subsequent positive effects on genomic stability and mitochondrial function. However, negative effects of CR on life and health span also have been reported (Szafranski and Mekhail 2014). This may be related to exhaustion of these metabolic sensors leading to deleterious effects in cell survival with age.
Although a connection between the AMPK pathway and ARHL has not yet been investigated in detail, the role of sirtuins in ARHL has gained particular relevance in recent years. The sirtuins (“silent mating-information regulators,” after their role in yeast) belong to a protein family with deacetylase or ADP-ribosyltransferase activities (Houtkooper et al. 2012). As previously mentioned, sirtuin enzymatic activity is linked to the cell energy state through NAD levels so that deacetylation by sirtuins is coupled to NAD hydrolysis. Several of the seven known mammalian sirtuins (Sirt1 to Sirt7) improve aging in mice (Houtkooper et al. 2012). While their actions are complex, many depend on improving genomic stability by epigenetic modifications of histones. It is interesting that a distinct role has been recognized for sirtuins in ARHL (Someya et al. 2010; Han and Someya 2013). CR limits oxidative damage to DNA in the cochlea and slows the progression of ARHL in wild-type mice. However, mice lacking the Sirt3 gene develop ARHL signs even under CR. It seems that Sirt3 directly deacetylates and activates mitochondrial isocitrate dehydrogenase 2 (Idh2), leading to increased NADPH levels and ultimately increased ratio of reduced/oxidized glutathione in mitochondria, thus protecting the auditory receptor from oxidative stress-induced cell death. If the effects of CR in ARHL may be mimicked and more finely controlled by direct (pharmacological) stimulation of sirtuins, this will open the door to new therapeutic targets for ARHL (Han and Someya 2013).
Accumulation of cellular damage caused by the “primary” aging factors previously discussed, along with reduced protective anti-damage mechanisms, can surpass the regulatory homeostatic capabilities of the cell. This leads to the age-damaged phenotype (Fig. 15.1), when there is also a decline in the regenerative potential of tissues. While mammalian inner ear sensory and neural cells show no regenerative potential, many other tissues of the inner ear do regenerate, and these cells are essential for tissue homeostasis (Menardo et al. 2012), although many details remain to be elucidated.
The previous paragraphs describing the role of genome damage and instability, altered epigenetic regulation, defective protein homeostasis, and reduced protective mechanisms give an idea of the general cellular aging landscape in which ARHL is embedded. Added to these specific mechanisms, there is a complex combination of genetic/intrinsic (e.g., sex, race, particular genes, etc.) and environmental/extrinsic (e.g., history of noise exposure, ototoxic drugs, etc.) factors that also contribute to the development of ARHL (Cruickshanks et al. 1998a, b; Agrawal et al. 2008; Lin et al. 2012; Marlenga et al. 2012; for review, see Yamasoba et al. 2013). In other words, systemic aging is a necessary but not sufficient condition for the development of ARHL. Along with contributions from sex and race, genetic risk factors (Lin et al. 2012; Marlenga et al. 2012) and predisposition to ARHL are reviewed in detail elsewhere (for reviews, see Yamasoba et al. 2013; Chap. 14 by RD Frisina and DR Frisina). It does not come as a surprise that many identified genetic variants and mutations related to susceptibility to ARHL involve genes related to antioxidation (see below) and also to glutamatergic neurotransmission (Sergeyenko et al. 2013; Yamasoba et al. 2013) and excitotoxicity (Pujol et al. 1991; Juiz et al. 1989). As far as environmental factors are concerned, the association of noise exposure and hearing loss is well known (see Chap. 7 by Altschuler and Dolan). In addition to simple additive relationships, noise-induced hearing loss (NIHL) that synergistically accelerates ARHL has more recently been recognized (Kujawa and Liberman 2006). Treatment with ototoxic drugs also results in hearing loss (see Chap. 10 by Rybak and Brenner, and Chap. 11 by Laurell and Pierre). To add further complexity, both noise damage and drug damage to the ear are also subject to genetic susceptibility (see Chap. 8 by Yamashita, Chap. 17 by Kohrman, and Chap. 18 by Green and Raphael). Thus, ARHL is a complex multifactorial disorder in which a lifelong accumulation of insults to the ear on a background of complex genetic predispositions and systemic aging leads to cell damage and hearing impairment (Gates and Mills 2005).
15.1.2 Histopathology of Age-Related Hearing Loss as a Mechanistic Framework
From a histological standpoint, ARHL is characterized by a progressive degeneration of hair cells starting at the basal end or high-frequency coding regions of the cochlea, atrophy and degeneration of the stria vascularis, and a variable degree of degeneration of the primary afferent SGN (Schuknecht and Gacek 1993). Therefore, cell populations in the cochlea involved directly in mechanotransduction (hair cells), in providing the electrochemical driving force for mechanotransduction (cells in the stria vascularis) or in transmitting and propagating transduced signals from the SGN to the central auditory pathway, may all degenerate and decrease in number and contribute to ARHL (Ohlemiller and Frisina 2008). Whether these universally acknowledged histopathological changes reflect independent, interdependent, or concurrent pathogenic events is an important question with impact on therapeutic strategies. A favored operational concept is that age-associated pathologies of the sensory epithelium, primary sensory neurons, or stria vascularis may arise and evolve independently, although at some point they may merge either by pathophysiological dependency on each other or simply by concurrence. This concept was elaborated by Schuknecht by correlating human patterns of hearing loss with the location of tissue damage and cell pathology in postmortem temporal bones. This seminal work led to the proposal of four major types of human presbycusis, according to the predominant histopathological pattern in relation to changes in auditory thresholds (Schucknecht 1955, 1964; Schuknecht and Gacek 1993; Ohlemiller and Frisina 2008; Schmiedt 2010).
In sensory presbycusis, the sensory epithelium is the site of primary damage particularly in high-frequency regions. A variable degree of progressive secondary SGN degeneration follows damage to the sensory lamina (in contrast to primary neural presbycusis). Age-related pathology primarily affecting hair cells may overlap with prolonged accumulation of noise or chemical damage to the auditory receptor making it difficult to separate contributions from preexisting lesions and aging. Therefore, to some extent, there may be a “pathogenic continuum” between accumulation of cell lesions from environmental factors and “true” age-related damage to sensory structures, with important consequences for prevention and treatment strategies.
In neural presbycusis, a specific primary loss of SGN with no damage to hair cells was proposed (Schucknecht 1955, 1964; Schuknecht and Gacek 1993; for recent reviews, see Ohlemiller and Frisina 2008; Schmiedt 2010). However, complex trophic relationships between SGNs and their target sensory epithelium challenge the notion of “pure” neural presbycusis. Certainly, there is evidence that SGN may experience progressive degeneration without detectable hair cell loss in young animals, and, conversely, they may survive for extended time periods after hair cell loss (Kujawa and Liberman 2006 ). Thus, SGN survival or death can be independent of hair cell targets in adulthood. Taken together, there may be a “pure” loss of SGN in neural presbycusis primarily due to aging mechanisms as recently shown in mice (see Boettcher et al. 1995; Schmiedt et al. 1996; Sergeyenko et al. 2013), or it may represent, again, a lifetime accumulation of insults, leading progressively to auditory neuropathy and overlapped with, and indistinguishable of, auditory aging.
In strial or metabolic presbycusis, the stria vascularis becomes atrophic and degenerates. The involvement of the stria vascularis in the pathogenesis of ARHL has attracted considerable attention, particularly in recent years. Schuknecht developed the concept that degeneration in the stria vascularis is a major contributor to hearing loss associated with the aging process (Schuknecht and Gacek 1993; Gates and Mills 2005; Schmiedt 2010). Strial pathology seems to be a common feature of ARHL in both humans and animal models. Actually it is the earliest identified pathological sign in the age-damaged cochlea, first seen in the third decade of life in humans, and it has the highest heritability index (Gates and Mills 2005; Ohlemiller 2009; Fetoni et al. 2011; Yamasoba et al. 2013). However, hearing loss characterized clinically as strial presbycusis sometimes includes loss of hairs cells in the basal turn of the cochlea as the most visible pathology. Some postulate that the hair cell loss could be the result of specific insults such as noise exposure, whereas the strial pathology is purely related to aging (Gates and Mills 2005).
Aged gerbils show a prominent degeneration and atrophy of the stria vascularis (Fetoni et al. 2011). They exhibit a progressive pathology of marginal and intermediate cells and of the Na+K+ATPase-positive fibrocytes in the spiral ligament (Spicer and Schulte 2002, 2005; Gates and Mills 2005). This age-dependent degenerative process involves loss of the expression of the Na+K+ATPase in the lateral wall and stria vascularis (Schulte and Schmiedt 1992). This ATPase regulates K+ and Na+ transport through the lateral wall and, as a consequence, the endocochlear potential (EP) (Spicer and Schulte 2005; Schmiedt 2010; for detailed discussion see Chap. 3 by Ohlemiller). Indeed, aged gerbils exhibit a progressive decline in EP and a disruption of ion homeostasis in the cochlea (Schulte and Schmiedt 1992; Schmiedt 1996, 2010). Thus, in aged animals, degeneration of the stria vascularis can lead to a progressive decline of the EP and impairment of the cochlear amplifier (Gates and Mills 2005; Schmiedt 2010).
Aged-related alterations in the microcirculation of the stria vascularis have been described in several experimental models (Shi 2011). The metabolic rate is high in the stria vascularis (Gates and Mills 2005; Fetoni et al. 2011; Böttger and Schacht 2013). Aged gerbils show atrophy and loss of strial capillaries that gradually expands from the apical to the middle region of the cochlea (Gratton and Schulte 1995). Age-related reduction of cochlear blood flow is exhibited by old gerbils specifically in the lateral wall (Prazma et al. 1990), and the EP decline observed in old gerbils and described above correlates with atrophic capillaries in the stria vascularis (Gratton et al. 1996). Further studies demonstrate age-dependent thickened basal membrane (BM) in 65–85 % with an increase of immunoglobulin and laminin deposition in strial capillaries (Sakaguchi et al. 1997a, b; Thomopoulos et al. 1997), suggesting BM alterations and age-dependent permeability in these vessels. Similarly, aged Fischer 344 rats show decrease in the vascularization of the stria vascularis (Buckiova et al. 2006) accompanied by age-dependent reduction of red blood cell velocity and increased capillary permeability (Seidman et al. 1996). Finally, in 18-month-old C57BL/6 J mice, a decreased expression of the specific endothelial marker VEGF in this tissue is described (Picciotti et al. 2004). These microvascular alterations contribute to the age-related pathology of the stria vascularis and the progressive worsening of ARHL.
Mechanical or conductive presbycusis is characterized by “stiffening” of the basilar membrane, although this remains unproven and under discussion (Gates and Mills 2005; Ohlemiller and Frisina 2008; Schmiedt 2010). However, presbycusis does not present in an isolated form of one of those described above. Instead it comprises a mixture of alterations that likely reflect the summation aging effects in many cell types (Schuknecht and Gacek 1993; Ohlemiller and Frisina 2008). In this regard, Schuknecht included a frequent form of presbycusis named mixed to point out that ARHL is often characterized by a broad cochlear degeneration due to coincident independent causes (Schuknecht and Gacek 1993). Actually, it has been recently proposed (Engle et al. 2013) that in the rhesus monkey, the accumulating number of cochlear pathologies correlates better with audiometric patterns than single pathologies. Finally, Schuknecht established the indeterminate presbycusis for those cases where no significant changes were observed in any cochlear structure and its audiogram differed from the conductive one (Schuknecht and Gacek 1993).
It is clear from the above that Schuknecht’s histopathological framework is limited and under continuous review. However, it still provides a valid context within which we may view new knowledge of ARHL mechanisms (Ohlemiller and Frisina 2008).
15.1.3 Mitochondrial Mutations and Dysfunction and Its Association with Age-Related Hearing Loss
Mitochondrial dysfunction is central to aging (López-Otín et al. 2013). Actually, altered mitochondrial function is of utmost relevance in ARHL, and it could be at the origin of all the histopathological types of presbycusis described by Schuknecht.
Mitochondria play a central role in the physiology of the cell as the primary source of ATP and are key players in many cellular processes, including calcium signaling and regulation of apoptotic cell death. Mitochondrial metabolism produces free radicals, including reactive oxygen species (ROS) and reactive nitrogen species (RNS) (for detailed discussion, see Chap. 2 by Leeuwenburgh). Both ROS and RNS have signaling roles under normal conditions but are toxic to the cell in excessive concentrations, leading to oxidative stress damage (Fig. 15.2) (Sena and Chandel 2012; Böttger and Schacht 2013). In this regard, mitochondrial dysfunction contributes to a large number of human age-related neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases, amyotrophic lateral sclerosis, and Huntington’s disease (Lin and Beal 2006) and damage attributed to aging (Lenaz et al. 2006; Lee and Wei 2012; Bratic and Larsson 2013). While there is much evidence supporting the idea that mutations in mitochondrial genomic DNA (mtDNA) and oxidative stress both contribute to aging (Lin and Beal 2006), recent evidence supports a primary role for mtDNA mutations in mammalian aging (Bratic and Larsson 2013; Liochev 2013; Viña et al. 2013). Thus, genetically modified mice undergoing progressive accumulation of mtDNA mutations exhibit an accelerated aging phenotype (Trifunovic et al. 2004; Kujoth et al. 2005). Further research demonstrated that these “mtDNA mutator mice” showed mitochondrial dysfunction unaccompanied by increased ROS production (Trifunovic et al. 2005), suggesting a direct link from the mtDNA mutation to mitochondrial dysfunction and the aging process. We will address the possible association between the mtDNA mutations and ARHL and then the role of ROS generation and oxidative stress in presbycusis.
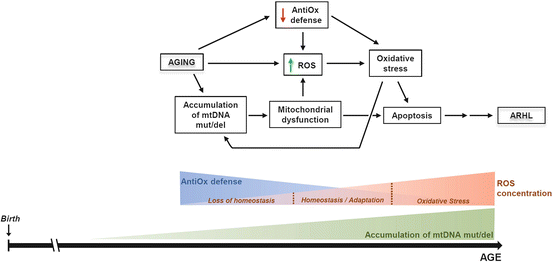
Fig. 15.2
Mitochondrial dysfunction in the pathogenesis of ARHL. Mitochondria have a central role in the pathogenesis of ARHL. During the aging process, many insults damage the cell and are progressively accumulated. In this regard, mitochondrial DNA (mtDNA) mutations and deletions (mut/del) are generated due to the “primary” aging factors and gradually accumulate. This leads to mitochondrial dysfunction that in turn contributes to an oxidative stress situation due to the additive effect of increased toxic levels of ROS and the progressive decline of antioxidant (AntiOx) enzymes that are observed with age in the cochlea. Both processes, mitochondrial dysfunction and oxidative stress, activate apoptosis pathways leading to death of specific cell populations in the auditory receptor contributing to and worsening the aging phenotype. Thus, in the mitochondria of the cochlear cells, there is a progressive accumulation of mtDNA mut/del that significantly contributes to the gradual increase in the ROS levels. Once the toxic level of ROS is reached, oxidative stress ensues, which is also enhanced by the gradual decrease of the antioxidant defense of the cell with age. All these factors will finally activate cell death pathways. The ROS concentration sketch is modified from Glasauer and Chandel (2013)
Mitochondrial genomic alterations and ARHL. As stated above, mtDNA deletions and mutations increase with age and also contribute to age-related pathology (Bratic and Larsson 2013; López-Otín et al. 2013). mtDNA mutations are at the origin of a large number of human genetic diseases, many of which include hearing loss (Fischel-Ghodsian 2003; Schon and Przedborski 2011). This suggests a key role for mtDNA mutations specifically in ARHL (Yamasoba et al. 2007). In this regard, it has been estimated that nearly 70 % of patients with mitochondrial genomic disorders also display sensorineural hearing loss (Gold and Rapin 1994; Seidman et al. 2004). In an animal model, 9-month-old “mtDNA mutator mice” showed a significantly greater loss of outer hair cells (OHCs) and SGNs in the basal turn of the cochlea, along with increased ABR threshold shifts, compared to age-matched controls (Kujoth et al. 2005). This suggests that the accumulation of mtDNA mutations in the cochlea as a function of aging may directly lead to hearing impairment attributed to age.
Bai et al. (1997) examined 34 human temporal bone samples, 17 of which belonged to people with ARHL. They found a 4977 base pair (bp) “common aging deletion,” which was significantly more frequent in cochlear tissues of ARHL patients than patients without ARHL. A similar 4834 bp common aging deletion was also found to accumulate with age and to be associated with progressive lower hearing sensitivity in Fischer rats (Seidman et al. 1997). Mutations in the mitochondrial cytochrome oxidase II gene in temporal bones from five ARHL patients have also been described, further supporting the likelihood that sporadic mtDNA mutations also contribute to ARHL (Fischel-Ghodsian et al. 1997). These observations were confirmed in humans by Dai et al. (2004), who found a high incidence of the mtDNA4977 deletion in the temporal bones of ARHL patients (17/34) compared to ears in the age-matched control group (4/19) and those in the young and middle-aged control group (0/14). Perhaps most compelling, a quantitative correlation was established between the level of the mtDNA4977 deletion in human temporal bone samples from ARHL patients and the severity of the hearing loss (Markaryan et al. 2009). These authors also observed a decrease in the expression of the mitochondrial cytochrome c oxidase subunit 3 (COX3) gene in SGNs and an increase in deletions different from the mtDNA common deletion in COX3-deficient SGNs (Markaryan et al. 2010). Taken together, these results are consistent with a causative relationship between the accumulation of mitochondrial genomic alterations with age, mitochondrial dysfunction, and the pathogenesis of ARHL (Fig. 15.2).
15.1.4 Mitochondria, Oxidative Stress, and Age-Related Hearing Loss
Because of the primarily energetic function of mitochondria, ROS are continuously produced in this organelle during the biochemical reactions of the electron transport chain. Superoxide is the primary radical, from which the rest of reactive species are generated through the activity of the cell antioxidant and detoxifying systems (Murphy 2009; Dröse and Brandt 2012). Redox regulatory mechanisms involve several antioxidant enzymes: superoxide dismutases (Cu/Zn-SOD and Mn-SOD), glutathione peroxidase (GPx), glutathione S-transferase (GST), and catalase (Balaban et al. 2005; Kowaltowski et al. 2009; Sena and Chandel 2012). Recent findings demonstrate that ROS also play essential roles in the physiology of the cell by acting in many cell signaling events (Finkel 2012; Ray et al. 2012; Sena and Chandel 2012) related to cellular homeostasis and its adaptation to stress situations (Sena and Chandel 2012). A currently accepted view is that oxidative stress implies an excessive concentration of ROS/RNS due to an imbalance between their generation and the inefficient capacity of the antioxidant systems to scavenge them (Fig. 15.2) (Lee and Wei 2012; Glasauer and Chandel 2013). It has been hypothesized that excessive production accumulates over time, damaging key molecules and cell structures (nuclear and mitochondrial DNA, membranes, and proteins) and resulting in tissue dysfunction during aging (Lee and Wei 2012). Since mitochondria are the major source of ROS (Balaban et al. 2005; Kowaltowski et al. 2009; Brand 2010), the primary effect of oxidative stress we expect would impact on mitochondria, leading to mitochondrial dysfunction with age as seen in several neurodegenerative diseases and other pathologies (Lin and Beal 2006; Sena and Chandel 2012).
Presbycusis is believed to reflect the progressive accumulation of metabolic alterations due in part to ROS/RNS and the progressive inefficient functioning of mitochondria with age on a background of unusually high metabolic demands in the inner ear tissues (Someya and Prolla 2010; Yamasoba et al. 2013). Although the stria vascularis has the highest aerobic metabolic rate of all cochlear structures (Böttger and Schacht 2013), iOHCs are also vulnerable to oxidative damage because of their high metabolic activity and lower content of antioxidants, like glutathione (Sha et al. 2001).
Recent evidence lends further support to the notion of oxidative stress as a contributing factor in the pathogenesis of ARHL (Someya and Prolla 2010; Yamasoba et al. 2013). There was a significant increase in oxidative stress markers (glutathione-conjugated proteins and 4-hydroxynonenal and 3-nitrotyrosine) and a decrease of the antioxidant scavengers AIF and SOD2 in the cochlea of aged CBA/J mice (Jiang et al. 2007), indicating an imbalance in the redox status of the cochlea with age. On the other hand, Sod1 deficiency in mice increased the age-related loss of both inner hair cells (IHCs) and OHCs following a base-to-apex gradient, suggesting a correlation between the SOD1 deficiency and the survival of the cochlear sensory cells (McFadden et al. 1999b). Further experiments correlated these hair cell losses with hearing impairment (measured by ABR thresholds) and showed age-related loss of SGN in the 13-month-old homozygous knockout mice (McFadden et al. 1999a). A study with the Sod1–null mice showed severe degeneration of SGN at 7–9 months of age, elevated threshold shifts in 12-month-old animals, and thickening of the stria vascularis at 15 months of age, a pattern of results suggesting early ARHL in Sod1 deficient mice (Keithley et al. 2005). In addition, overexpression of mitochondrially targeted catalase in C57BL/6 J mice delayed the onset of ARHL in mice by delaying hair cell loss (Someya et al. 2009). Finally, a moderate age-related increase in GPx activity has been described in the spiral ligament and stria vascularis in 24-month-old Fisher 344 rats (Coling et al. 2009). Taken together, these results indicate that imbalances in the cellular antioxidant machinery lead to early onset of ARHL and point to oxidative stress as an important contributing factor in its pathogenesis (Fig. 15.2).
If oxidative stress plays a causative role in the pathogenesis of ARHL, it is likely that priming antioxidant mechanisms may reduce cochlear damage and delay onset of pathological changes. However, in contrast to noise-induced and drug-induced hearing loss, where a causal relationship between oxidative stress blocking and improvement in hearing impairment is reasonably well established, in the case of ARHL the link is weaker (Böttger and Schacht 2013). Although many studies have shown effects of antioxidant administration against ARHL, others have not. Seidman (2000) studied the independent effects of several antioxidants (vitamin E, vitamin C, melatonin, and lazaroid) in Fischer 344 rats maintained on lifelong (average, 25 months) supplemented diets. They observed improved auditory thresholds and fewer mtDNA deletions in antioxidant-treated animals, with some agents being more protective than others (see also Chap. 4 by Seidman and Shirwany). Similar results were obtained with the oral administration of lecithin to 18- to 20-month-old Fischer 344 rats for 6 months (Seidman et al. 2002). Heman-Ackah et al. (2010) fed C57BL/6 mice with a combination of six antioxidant agents (l-cysteine-glutathione mixed disulfide, ribose cysteine, NW-nitro-l-arginine methyl ester, folate, vitamin C, and vitamin B12) and observed a significant decrease in threshold shifts in 12-month-old antioxidant-treated animals. Moreover, in C57BL/6 mice supplemented with the mitochondrial antioxidants α-lipoic acid and coenzyme Q10, a reversal in ARHL was demonstrated, with lower hair cell and SGN cell death (Someya et al. 2009). However, 15-, 18-, or 24-month-old Fischer 344 rats treated with l-carnitine with different methods of administration did not show improvement in progression of ARHL (Bielefeld et al. 2008), and 10-month-old CBA/J mice supplemented with an antioxidant-enriched diet composed of α-lipoic acid, vitamin A, vitamin C, vitamin E, and l-carnitine showed no delay in the onset of presbycusis or loss of hair cells or SGN at 24 months of age (Sha et al. 2012). Additional discussion of therapeutics administered to animal subjects is found in Chap. 16 by Yamasoba.
Taken together, these findings suggest that although oxidative stress is a contributing factor to the pathogenesis of ARHL, it is currently difficult to establish it as a causative factor, without further investigation. In this regard, our lab is studying the otoprotective mechanisms of a combination of antioxidants (vitamin A, vitamin C, and vitamin E) plus magnesium (Mg++). Mg++ has been demonstrated to increase inner ear blood flow (Scheibe et al. 2000; Haupt and Scheibe 2002), and its efficacy in attenuating NIHL has been demonstrated (Sendowski 2006). The ACEMg treatment was administered to guinea pigs (Le Prell et al. 2007) and CBA/J mice (Le Prell et al. 2011) showing reduced permanent threshold shifts after NIHL and also reduced threshold shift after gentamicin ototoxicity (Le Prell et al. 2014). Studies in humans indicate that regular dietary intake of antioxidants correlates with lower risks of hearing loss (Durga et al. 2007; Shargorodsky et al. 2010; Choi et al. 2014). Chapter 6 by Spankovich discusses epidemiological data on the role of nutrients in hearing loss prevention in humans.
15.1.5 Apoptosis Regulation and Age-Related Hearing Loss
Several animal models have been used to study the role of apoptosis in the pathogenesis of ARHL (Someya and Prolla 2010; Fetoni et al. 2011). Experiments with old gerbils and mice show that in the aged cochlea, there are various cell types that undergo programmed cell death, such as IHCs and OHCs and supporting cells in the organ of Corti and fibrocytes in the spiral ligament (Usami et al. 1997; Spicer and Schulte 2002). Similarly, aged Fischer 344 rats exhibit increased TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling)-positive cells in both the marginal and basal cell layers of the stria vascularis and cells of the spiral ligament. Apoptosis results in a detachment of both layers. This correlates with altered otoacoustic emissions (Buckiova et al. 2007).
The molecular mechanism involved in such apoptotic death has been extensively studied. Experiments with aging gerbils and rats show downregulation of the anti-apoptotic Bcl family proteins Bcl-2 and Bcl-xL and the activation of pro-apoptotic proteins Bax and Caspase 3 (Alam et al. 2001; Nevado et al. 2006) leading to the hypothesis that the intrinsic pathway of apoptosis may mediate this aged-related cell death. In Fischer 344 rats, Hu et al. (2008) established a temporal sequence of molecular events associated with cell death molecular. Bax expression, release of mitochondrial cytochrome c, and DNA fragmentation take place before the nuclear condensation, followed by activation of caspases 3 and 9, continuing to the degradation of F-actin. On the other hand, gene expression studies in mice demonstrate age-dependent changes of apoptosis-related genes in the cochlea of aging animals (Tadros et al. 2008) and the central role of the mitochondrial pathway of apoptosis via the Bak protein as the key player in this process (Someya et al. 2009). The role of these pathways to cell death is brought together by Sha et al. (2009) who have shown that multiple cell signaling and cell death pathways (both the intrinsic and extrinsic apoptosis and also necrosis) are progressively activated in the cochlea of CBA/J mice with age. Finally, the overexpression of the anti-apoptotic protein XIAP (X-linked inhibitor of apoptosis protein) delays the progression of ARHL and decreases hair cell death in C57BL/6 J mice (Wang et al. 2010).
15.1.6 Cellular Mechanisms of Age-Related Hearing Loss in the Central Auditory Pathways
An understanding of ARHL and possible intervention strategies is incomplete without consideration of cellular and molecular changes associated with auditory aging in the central auditory pathways and their functional consequences. There has been comparatively less attention given to central mechanisms contributing to ARHL, and we strongly believe there is a need for a more integrated view of ARHL, combining peripheral and central mechanisms. Such an approach will not only accelerate understanding of ARHL but will also lead to more efficient management and therapeutic strategies through timely targeting of peripheral and central pathogenic mechanisms (Parham et al. 2011; Gates 2012; Kim and Chung 2013).
Defective processing by central auditory neurons and circuits in ARHL has a dual origin. One is declining peripheral input from the auditory receptor with age. This leads to diminished activity in central auditory neurons which is visible in auditory brainstem response evoked potentials (Fig. 15.3), with consequences for signal propagation and transmission, as well as trophic maintenance of nerve cells. The second is the aging process in the brain itself, which also leads to altered neuronal circuit function, although probably not through identical mechanisms. Both may overlap considerably and even be part of a pathogenic continuum in ARHL. Thus, a major challenge for research on central mechanisms of ARHL is to distinguish between relative contributions and impact of age-declining peripheral input versus direct effects of aging on the central auditory system and to establish their pathogenic sequences and timelines. Sorting the dual origins of the central causes and consequences of ARHL and, equally important, understanding how they interact will have an impact on future therapeutic strategies.
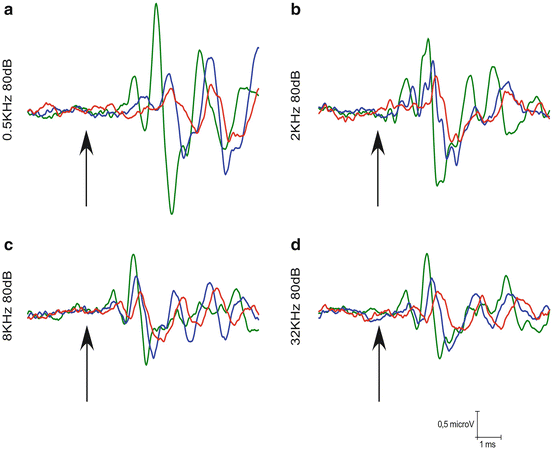
Fig. 15.3
Changes in auditory brainstem responses with age in the rat. An example of ABR recordings in Wistar rats at 6- to 8-month-old (green), 12- to 14-month-old (blue), and 18- to 20-month-old (red). Wave amplitudes decrease at the different frequencies evaluated 0.5 (A), 2 (B), 8 (C), or 32 (D) kHz, and latencies increase as the age of the rats increase. Arrows indicate the stimulus onset. Stimulus intensity = 80 dB SPL
Animal models, in particular rodent strains with various onset, severity, and patterns of hearing loss, have been very useful to dissect out the contributions to central ARHL mechanisms of age-diminished peripheral input versus central aging (see Ohlemiller and Frisina 2008 for review and their chapters in this book). There is evidence that the progression of age-related loss of synaptic input from the auditory receptor affects neuronal survival, trophic support, and signal propagation and transmission (Fig. 15.3), with greatest severity of structural changes in caudal nuclei of the auditory brainstem. Neurons in the cochlear nucleus (CN) are primary recipients of synaptic endings from auditory nerve axons, and therefore the first to “sense” diminished peripheral input. There is an overall reduction of neuron number, packing density, and reduced cell size, distributed tonotopically and consistent with the time course and progression rate of the peripheral hearing loss (Willott et al. 1987, 1992). Early loss of high-frequency hearing in the C57 mouse strain (Willott et al. 1987) leads to early and stable structural changes in the CN. Delayed hearing loss, as in the CBA mouse (Willott et al. 1987), corresponds with delayed structural changes in aged CN neurons of similar nature. It is interesting that these peripherally driven effects on neuronal structure do not affect uniformly all CN neuronal populations, with octopus cells in the posteroventral CN being particularly resilient to limited auditory nerve input (Willott and Bross 1990). The type of neuron and its pattern of synaptic inputs seem to determine neuronal survival and trophic maintenance in the presence of limited peripheral input with age. However, it is also interesting to note that mouse strains (Willott et al. 1987) or those humans having a significantly larger number of neurons in the CN (Hinojosa and Nelson 2011) seem to be more prone to neuronal loss attributable to presbycusis, suggesting an added genetic component to peripherally driven neuronal loss. In the auditory pathway rostral to the CN, neuronal loss attributable to auditory aging is much more limited or not present (Shim et al. 2012; Willott et al. 1994). Neuronal body size diminishes or remains stable “upstream” of the CN, at least in the inferior colliculus (IC) (Willott et al. 1994; Kazee et al. 1995). Therefore, it seems that trophic support or survival of many CN neuron types is severely compromised by age-dependent reduction in direct peripheral inputs carried by the auditory nerve. However, at more rostral levels of the auditory pathway, beyond the primary inputs, diminished peripheral activity has much more limited impact on net cell survival, supporting the view that trophic signals differ at different levels of the auditory pathway, with possible consequences for the aging process.
As a consequence of damage to neuronal cell bodies, particularly in the CN, axonal degeneration is expected. However, the extent and rate of age-related degeneration of axons traveling in auditory tracts is unclear. Axonal connections from the CN to the inferior colliculus (IC) are not significantly reduced in mice with early signs of ARHL at any age (C57, Willott et al. 1985). However, in mice with mild ARHL, connections from the CN to the IC are significantly reduced with age (CBA; Frisina and Walton 2001, 2006). The number of fibers in the human lateral lemniscus is also reduced at advanced ages (Ferraro and Minckler 1977, cited in Ohlemiller and Frisina 2008). Regardless the extent and rate of axonal degeneration, connections from the CN to more rostral auditory nuclei carry degraded signals (reviewed in Frisina and Walton 2006) as a consequence of aging (Fig. 15.3). Among other things, as a consequence of high-frequency loss, there are rearrangements in the tonotopic map of more rostral nuclei, like the IC (Willott et al. 1991), with high-frequency regions becoming more sensitive to lower-frequency sounds. The nature of this peripherally induced “rewiring” is unknown, but it could add to limited frequency discrimination. In the auditory cortex, plastic reorganizations of the tonotopic map also take place after age-related high-frequency hearing loss, with low frequencies expanding its representation to higher-frequency regions (Willott et al. 1993). The reorganization in connectivity underlying such age-related auditory cortex plasticity is not known. Such plastic reorganizations in the auditory midbrain and forebrain do not seem to take place in rodent models aging with good hearing preservation (Willott et al. 1993).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


