Chapter 18 Cellular Growth Control and Cancer
The cell cycle is controlled at two checkpoints
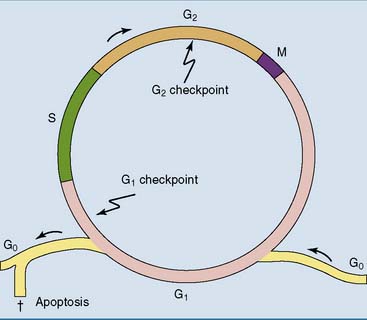
Under the microscope, only two phases of the cell cycle can be distinguished in dividing cells: interphase and mitosis (Fig. 18.1). Mitosis, which lasts between 1 and 4 hours, is the stage of cell division. All of the rest is interphase. Chromosomes are visible as distinct entities only during mitosis, when the DNA is packaged for relocation into the daughter cells. During interphase, there is only dispersed chromatin all over the nucleus.
Cells can be grown in culture
Cyclins play key roles in cell cycle control
Cell cycle progression depends on the phosphorylation of multiple proteins by the cyclin-dependent kinases (Cdks) in the nucleus and their regulatory subunits, the cyclins. The Cdks are present more or less at all times, but most of the cyclins come and go with the phases of the cell cycle (Fig. 18.2).
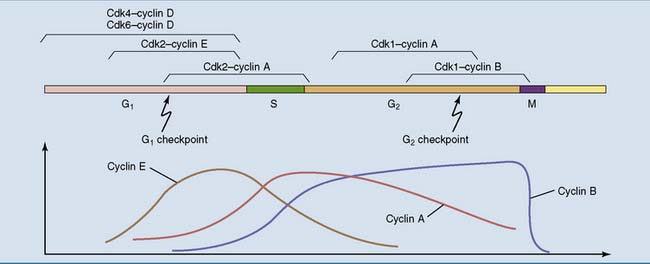
Figure 18.2 Cyclins and cyclin-dependent kinases (Cdks) during the cell cycle. Most cyclins are short lived, and their levels fluctuate with the stages of the cell cycle. The cyclin-dependent kinases and cyclin D, on the other hand, are present throughout the cell cycle. G1, S, G2, and M are the stages of the cell cycle (see Fig. 18.1).
Retinoblastoma protein guards the G1 checkpoint
Progression through the cell cycle and DNA synthesis require the transcription factor E2F, which regulates more than 500 genes. It activates the transcription of genes for cyclins D1 (there are three closely related D cyclins), E, A, and B, Cdk1, thymidylate synthetase, dihydrofolate reductase, DNA polymerase α, topoisomerase II, the clamp protein PCNA (see Chapter 7), and the proto-oncogenes MYC and MYCL1 (which code for the transcription factors c-Myc and N-Myc, respectively).
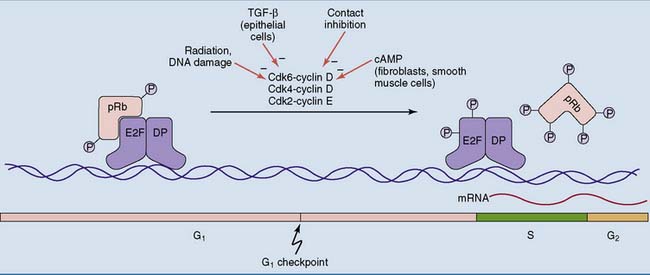
In quiescent cells, E2F is prevented from activating the transcription of these genes by the retinoblastoma protein (pRb), which is encoded by the RB1 gene (Fig. 18.3). Throughout G0 and early G1, pRb is tightly bound to E2F. It prevents gene expression by masking the transcriptional activation domain of E2F and by recruiting a histone deacetylase. At the G1 checkpoint, however, both pRb and E2F become phosphorylated by the kinase complexes of cyclins D and E. The phosphorylated pRb falls off the transcription factor, and the genes can be transcribed.
Cell proliferation is triggered by mitogens
Mitogenic stimuli can be provided by the extracellular matrix. The integrins in focal adhesions (see Chapter 13) not only mediate adhesion to extracellular matrix components but also provide an assembly point for signaling molecules. Cell-matrix contacts tend to be mitogenic, but cell-cell contacts usually are antimitogenic.
Mitogens regulate gene expression
Mitogens push cells through the G1 checkpoint. Figure 18.4 shows two mitogenic signaling cascades that are triggered by autophosphorylated growth factor receptors and activate the nuclear cyclin D-Cdk complexes.
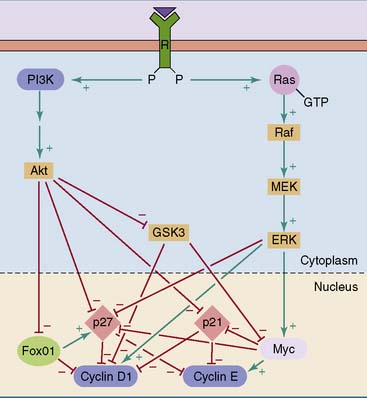
One of these cascades signals through phosphoinositide 3–kinase (PI3K) and protein kinase B (PKB, or Akt) (see Chapter 17). PKB phosphorylates and thereby inhibits another protein kinase, glycogen synthase kinase 3 (GSK3). GSK3 inhibits the expression of cyclin D1 by phosphorylating transcriptional regulators bound to the promoter of the cyclin D1 gene. By inhibiting these inhibitory phosphorylations, PKB stimulates the expression of the cyclin D1 gene.
The other mitogenic cascade shown in Figure 18.4 is the mitogen-activated protein (MAP) kinase cascade. It starts with activation of the small G protein Ras at the cytoplasmic surface of the plasma membrane (see Chapter 17). Three isoforms of Ras occur in human tissues.
Negative controls on mitogenic signaling include the dephosphorylation of proteins by protein phosphatases at all levels, from the autophosphorylated growth factor receptors to the phosphorylated transcription factors. Another negative control is the hydrolysis of its bound GTP by the Ras protein. The GTPase activity of Ras is stimulated by regulatory proteins, including neurofibromin (Clinical Example 18.2).
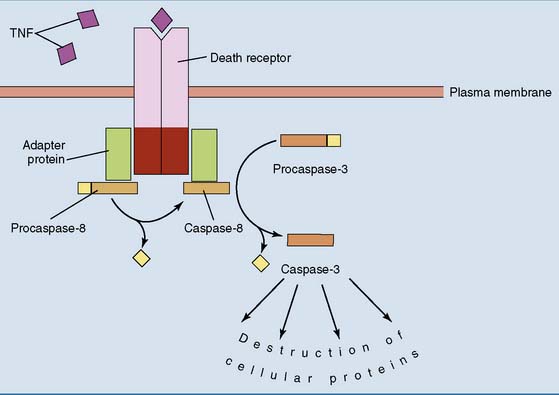
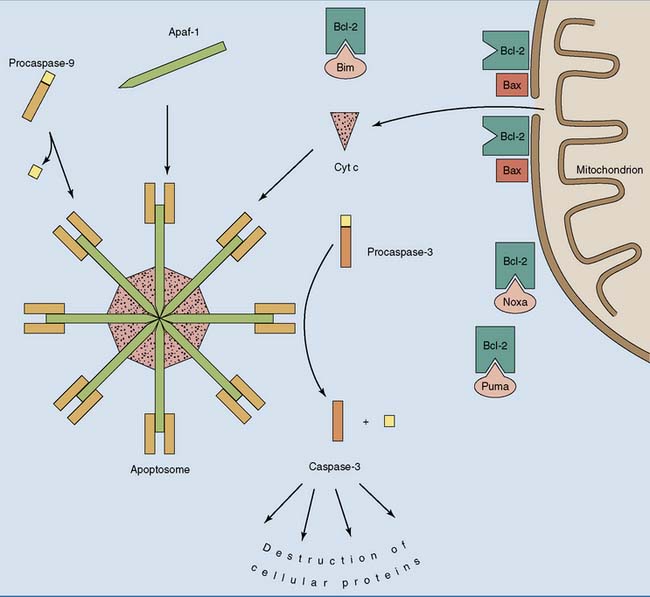
Cells can commit suicide
Cancers are monoclonal in origin
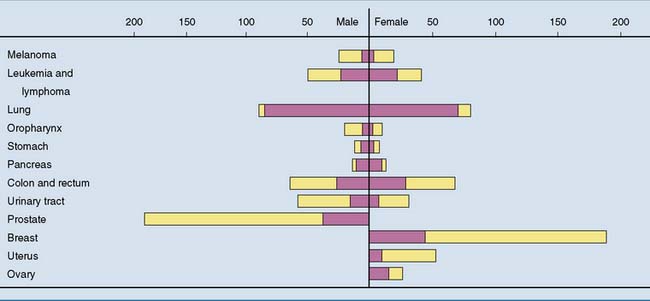
Cancer cells, however, have the cellular equivalent of antisocial personality disorder. A cancer cell arises when a somatic mutation creates a “selfish gene” that causes the cell to proliferate without regard for the greater good of the organism. This single abnormal cell grows into a cell mass called a neoplasm or tumor. Most tumors are monoclonal in origin. This means that all tumor cells are derived from a single abnormal ancestor. Benign tumors limit their growth without doing much harm, but malignant tumors, commonly called cancer, kill the organism. Cancer causes more than 20% of all deaths in industrialized countries. Figure 18.7 shows the incidence of various kinds of cancer in the United States.
Cancer is caused by activation of growth-promoting genes and inactivation of growth-inhibiting genes
Some gene products, including growth factors, growth factor receptors, components of mitogenic signaling cascades, and the G1 cyclins, promote cell proliferation. Others, including cyclin-Cdk inhibitors and pRb, are inhibitory. Therefore two types of mutation can favor mitosis (Fig. 18.8):
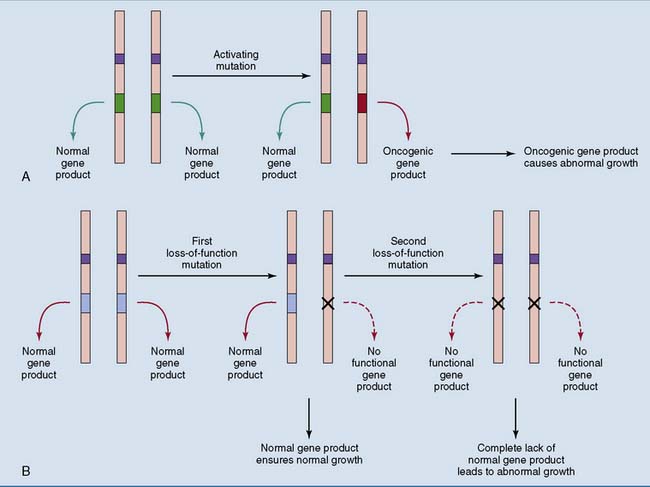
 ) are growth-stimulating proteins. A single activating mutation (“gain-of-function” mutation) is sufficient to produce abnormal cell growth. B, Products of tumor suppressor genes (
) are growth-stimulating proteins. A single activating mutation (“gain-of-function” mutation) is sufficient to produce abnormal cell growth. B, Products of tumor suppressor genes ( ) are growth-inhibiting proteins. Two inactivating mutations (“loss-of-function” mutations) are necessary to produce abnormal cell growth.
) are growth-inhibiting proteins. Two inactivating mutations (“loss-of-function” mutations) are necessary to produce abnormal cell growth.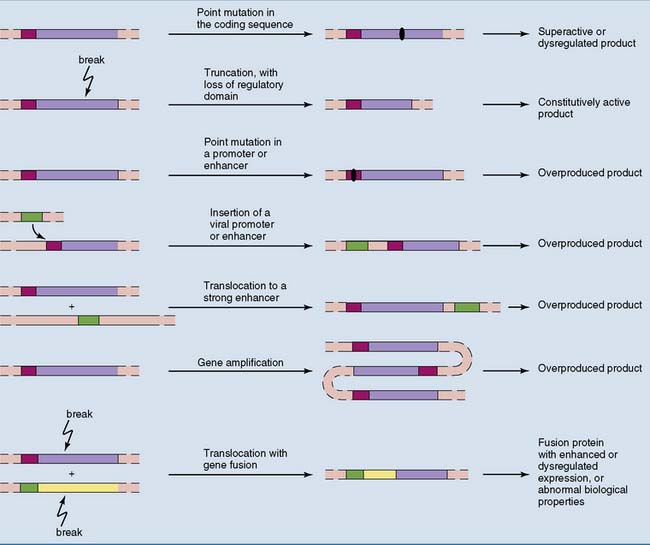
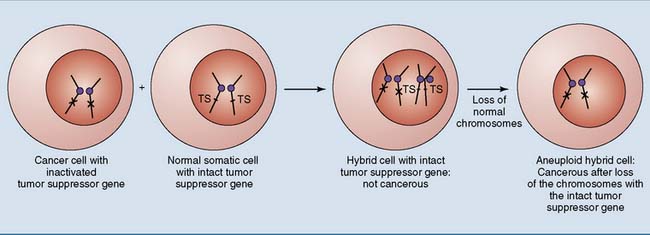
Inactivation of tumor suppressor genes is even more important than activation of cellular proto-oncogenes in most spontaneous cancers. This can be demonstrated in cultured cells (Fig. 18.10). When a cancer cell is fused with a normal cell, the resulting hybrid cell grows like a normal cell because the intact tumor suppressor gene from the normal cell produces the tumor-suppressing protein. If malignant transformation were caused by dominantly acting oncogenes, the hybrid cells would grow like cancer cells.