Fig. 1.1
Schematic showing stages of preimplantation development, the derivation of early cell lineages and morphogenesis of the embryo with the key steps and consequences of intrinsic cell-cell interactions occurring at 8-cell compaction and later blastocyst formation highlighted
1.2.1 Cell Signalling at Fertilisation and Activation of the Development Programme
Fertilisation not only achieves a diploid bi-allelic genome to drive the developmental programme but also represents the first and most crucial cell-cell signalling event that prepares the egg for executing that programme. Sperm-derived phospholipase C-zeta (PLCζ) enters the egg at gamete fusion and initiates, through the phosphoinositide signalling pathway, a series of Ca2+ oscillations to activate the fertilised egg. The PLCζ hydrolyses a pool of phosphatidylinositol 4,5-bisphosphate (PIP2) that appears to be derived from intracellular vesicles rather than the more usual plasma membrane complement to drive production of inositol 1,4,5-triphosphate (IP3) (Swann and Lai 2013). IP3 binds to IP3 receptors in cortical endoplasmic reticulum to release stored Ca2+ to initiate the oscillations. Downstream targets of the sperm-mediated signal include (i) exocytosis of cortical granules to modulate the composition of the zona pellucida and prevent polyspermy; (ii) reactivation of cell cycling following meiotic arrest leading to the formation of the second polar body and distinct cytoplasmic male and female pronuclei; (iii) activation of the new embryonic genome to drive the embryonic programme of development (White et al. 2010; Nomikos et al. 2012; Dehapiot et al. 2013; Swann and Lai 2013).
The pathway from fertilisation signalling to the activation of the development programme leading to blastocyst formation is now thought to comprise both maternal and embryonic control mechanisms. Recent evidence indicates stored maternal factors encoded during oogenesis by the maternal genome facilitate embryonic genome activation (EGA) following the fertilisation signal. These factors contribute especially to the processing of the male genome, degradation of unnecessary maternal RNAs and proteins to allow for EGA, early cleavage divisions and initial steps in the emergence of distinct cell lineages (Li et al. 2010). Perhaps the key factors involved in maternal-embryonic transition regulating blastocyst morphogenesis comprise transcription factors that tend to stabilise early cell lineages formed through cell-cell signalling and resultant cell asymmetry and differentiative divisions. These cell signalling events are therefore discussed first before returning to transcription factor stabilisation of lineages later.
1.2.2 Cell Signalling at Compaction
The fertilisation signal and activation of the development programme leads to the newly-formed embryo initiating cell cycling after the period of meiotic arrest following ovulation. Thus, cleavage to 2-, 4- and 8-cell stages in the mouse occurs in the absence of an increase in biomass over the subsequent 2.5 days with cycles being asynchronous, and with blastomeres being clearly distinct and similarly sized (Fig. 1.1). Compaction in the 8-cell mouse embryo represents a morphological transition when these early blastomeres first demonstrate cell-cell signalling and interactions that are essential for morphogenesis of the blastocyst over the next 1–2 days. As the name implies, compaction comprises the activation of cell-cell adhesion and the formation of adherens junctions between individual blastomeres such that their outlines are no longer distinct and a spherical ball of cells, the morula, is formed. Intercellular adhesion is mediated mainly through the Ca2+-dependent E-cadherin-catenin complex (Vestweber and Kemler 1985; Johnson et al. 1986) but also through the co-localising vezatin adhesion complex (Hyenne et al. 2005), the Ca2+ -independent immunoglobulin family nectin complex (Thomas et al. 2004) and the epithin membrane serine protease (Khang et al. 2005). Close cell-cell adhesion at compaction is accompanied by restructuring of the membrane cytoskeleton involving nucleation and formation of actin filaments (Sun et al. 2013) facilitated by the suppression in expression of the actin filament depolymerising protein, cofilin-1 (Ma et al. 2009). Recently, live cell imaging has revealed an elaborate system of E-cadherin- and myosin-dependent filopodia drawing neighbouring cells together to control cell shape changes at compaction (Fierro-Gonzalez et al. 2013).
In addition to close cell-cell adhesion, compaction also involves the onset of epithelial biogenesis within the embryo with each blastomere changing from a non-polar to polar phenotype comprising a clear apical domain (facing outwards, cell contact-free, non-adhesive and microvilli-rich) and a basolateral domain (facing inwards, adhesive, cell contact site, microvilli-poor) (Fleming et al. 2001) (Fig. 1.1). The polarised organisation of blastomere surfaces is accompanied by restructuring of the cytoplasm with apico-basal polarity evident especially in more apical cytoskeletal actin and microtubules (Johnson and Maro 1984; Houliston et al. 1987) which facilitate relocation of vesicular endocytic organelles to the apical cytoplasm and initiation of an apico-basal polarity in endocytosis activity (Fleming and Pickering 1985; Maro et al. 1985; Fleming et al. 1986). The integrated cell adhesion and cell polarisation events at compaction form the basis of continued epithelial differentiation occurring on the surface of the embryo during subsequent 16- and 32-cell stages (~ 24 h) culminating in TE formation and blastocyst morphogenesis. In addition, asymmetric cell divisions of polarised 8- and 16-cell stage blastomeres generate the internal ICM lineage of the blastocyst (Fig. 1.1).
The timing of embryo compaction does not appear to be mediated through the expression of one or more adherens junction components acting as limiting factor but rather as a result of a complex cell-cell signalling event which is not yet clearly understood. Thus, expression of E-cadherin, and cytoplasmic domain intermediates (α-catenin, β-catenin, plakoglobin) can be detected in non-functional forms during earlier cleavage (Ohsugi et al. 1996). Functional activation of adhesion appears to be restricted until the 8-cell stage by a rapidly turning over inhibitor that allows for a coordination of compaction across the embryo (Levy et al. 1986). Experimental evidence suggests the activation of compaction likely comprises several interacting signalling components. For example, chemical activation and/or inhibition of protein kinase C (PKC) has been shown to promote or inhibit compaction, respectively. Moreover, during compaction, the PKCα isoform relocates from cytoplasmic to cell-cell contact sites coinciding with phosphorylation of β-catenin on serine/threonine residues and its recruitment from the soluble cytosolic fraction to the insoluble cytoskeletal pool (Pauken and Capco 1999, 2000). In addition, dephosphorylation of β-catenin at tyrosine sites has been detected around the time of compaction (Ohsugi et al. 1999).
The cell-cell signalling process of compaction also propagates away from the intercellular contact site to coordinate cell polarisation both within the deeper cytoplasm and at the contact-free apical surface. Inhibition of E-cadherin adhesion prior to compaction does not block cell polarity but causes a delay in its inception and disturbs its orientation (Johnson et al. 1986). Apical–basal epithelial polarity is controlled by several conserved polarity factors comprising the Par3/Par6/aPKC complex that regulates asymmetry in the distribution of membrane and underlying cytoskeleton organization and cytoplasmic constituents (Chen and Zhang 2013). Thus, the outward-facing apical pole of microvilli that forms on 8-cell blastomeres at compaction assembles components of the Par3/Par6/aPKC complex and disruption in their expression affects blastocyst morphogenesis and TE:ICM lineage allocation (Thomas et al. 2004; Plusa et al. 2005; Vinot et al. 2005). Signalling through Rho-GTPases is essential for cell polarisation to occur at compaction (Clayton et al. 1999) and contributes to the apical localisation of the Par3/Par6/aPKC complex (Liu et al. 2013). Ezrin, an actin-binding member of the ERM protein family, accumulates at the apical microvillous pole also under Rho family control and is directly involved in pole formation following activation through phosphorylation (Dard et al. 2004; Liu et al. 2013). Experimental evidence suggests that ezrin phosphorylation is induced through the PKC signalling pathway that activates compaction and also by the apical aPKC within the Par3/Par6/aPKC complex (Liu et al. 2013). Cell polarisation at compaction must coordinate activity across each cell of the embryo , linking the apical polarity complex with cytoplasmic re-organisation including apical accumulation of actin and microtubule cytoskeleton and endocytic vesicles (see above). The signalling protein, Prickle2, acts to integrate cell polarity through the Par3/Par6/aPKC complex with microtubule polarity within the cytoplasm (Tao et al. 2012).
1.2.3 Cell Signalling and Blastocyst Formation
Cell signalling and resultant cell polarity established at compaction becomes further elaborated during the 16- and early 32-cell stages in outer blastomeres to coordinate TE differentiation and blastocyst morphogenesis. The epithelial polarised phenotype matures during this period with respect to membrane, cytoskeletal and organelle constituents leading to a functional transporting epithelium capable of generating the blastocoel cavity (Fleming et al. 2001).
One major elaboration of cell polarity dependent upon cell signalling interactions is the construction of tight junctions at the apicolateral border between neighbouring outer polar cells forming the TE lineage. Tight junction membrane and cytoplasmic domain proteins assemble at this site in a sequential process that starts at compaction and is only completed just prior to blastocoel accumulation at the 32-cell stage (Eckert and Fleming 2008). Delayed transcription and translation of ZO-1 α+ isoform appears critical in regulating final delivery of membrane constituents, intercellular sealing and completion of tight junction assembly (Sheth et al. 1997). Indeed, siRNA knock down of ZO-1 but not the related cytoplasmic domain scaffold protein, ZO-2, inhibits blastocyst formation (Sheth et al. 2008; Wang et al. 2008). A further elaboration of the polarised epithelial phenotype necessary for blastocyst formation is the accumulation of functional basolateral membrane Na+K+-ATPase to drive apicobasal vectorial transport. A similar sequential pattern of expression of Na+K+-ATPase subunits regulates functional activation to the 32-cell stage mediated through expression of the β1-subunit (MacPhee et al. 2000; Madan et al. 2007).
Cell signalling regulating TE epithelial differentiation and blastocyst formation appears upstream of both tight junction biogenesis and Na+K+-ATPase activity. Thus, SRC family kinases are required for development and maintenance of tight junction integrity and specific SRK inhibitors induce tight junction permeability and blastocyst collapse (Giannatselis et al. 2011). p38 MAPK regulates filamentous actin dynamics during the morula stage (Paliga et al. 2005) and controls tight junction permeability and blastocyst expansion (Bell and Watson 2013).
1.2.4 Cell Signalling and the Stabilisation of Emergent Cell Lineages
We have seen that cell signalling mediating compaction leads to cell polarity and subsequent formation, through differentiative divisions at 8- and 16-cell stages, of outer TE and inner ICM cell lineages of the blastocyst (Fig. 1.1). These cell types are stabilised through expression of lineage-specific transcription factors , notably Cdx2 (Caudal-related homeobox 2) in maintaining TE epithelial differentiation and the pluripotency factors Oct4, Sox2 and Nanog in coordinating ICM viability (Cockburn and Rossant 2010; Miyanari and Torres-Padilla 2010; Bruce 2013; Saiz and Plusa 2013). These distinct transcription factors first show co-expression throughout the embryo but cell interactions and reciprocal inhibition lead to a lineage-specific expression profile (Niwa et al. 2005). The TEA domain family transcription factor, Tead4, acts upstream of Cdx2 to promote it and other transcription factors controlling TE fate (Nishioka et al. 2008). Cell interactions and transduction cascades also refine the localisation of Cdx2 to the outer cell layer through manipulation of the Hippo signalling pathway (Nishioka et al. 2009). Thus, Tead4 and its co-activator Yap enter outer cell nuclei to activate Cdx2 transcription. However, in inside cells, Yap is phosphorylated by the Hippo signalling kinase, Lats, leading to its cytoplasmic degradation and suppression of Tead4-mediated Cdx2 transcription. The difference in the number of contact domains between outer and inner cells may be sufficient to explain the difference in Hippo activity. Alternatively, signals from the apical polarity factors discussed above (Par3/Par6/aPKC) or related components (e.g. ezrin) in outside cells may act to suppress Hippo signalling here (Nishioka et al. 2009).
The cell signalling processes discussed above leading to emergence of TE and ICM cell types also play a part in coordinating the appropriate levels of expression of lineage-specific transcription factors . Thus, mRNA for Cdx2 accumulates in the apical cytoplasm of polarised 8-cell blastomeres at compaction, mediated through both actin and microtubule dynamics, and leads to its preferential inheritance into outer daughter blastomeres of the TE lineage (Skamagki et al. 2013). Upregulation of Cdx2 in outer cells following cell polarisation thereby maintains the TE differentiation programme (Ralston and Rossant 2008). However, an earlier, maternally-expressed pool of Cdx2 may further contribute to the establishment of cell polarity prior to the zygotic pool formed following EGA by acting to commit outer cells to the TE lineage (Jedrusik et al. 2010). The integration cell-cell signalling activities with the regulation of transcription factor cascades will continue to be an important focus for research in cell lineage diversification and blastocyst morphogenesis in the future.
1.3 Extrinsic Signalling Pathways
In vivo, the oviductal and uterine milieux contain all the necessary components to support early pregnancy . This includes nutrients and signalling molecules such as hormones, growth factors and cytokines collectively influencing blastocyst morphogenesis. A variety of communication processes within the post-conception maternal tract have been linked to embryo developmental progression, differentiation, implantation or immune system modulation which are commonly mediated by growth factors and cytokines (Hardy and Spanos 2002; Armant 2005; Robertson 2007; Koga and Mor 2008; Guzeloglu-Kayisli et al. 2009; Douglas 2011). Other chapters will discuss embryonic stress responses, calcium signalling , signalling via growth factors and cytokines, mechanisms surrounding glucose homeostasis, and the role of amino acids in early development . Here, we will discuss our current understanding of some general mechanisms the embryo may utilise to sense and respond to its nutritional environment during blastocyst morphogenesis with focus on maternal body physiology (Fig. 1.2).
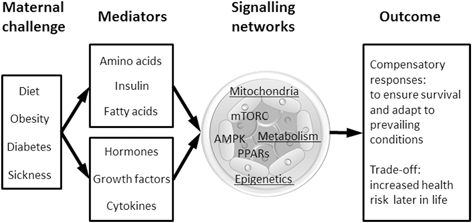
Fig. 1.2
Schematic showing the range and effects of extrinsic factors and embryo signalling pathways together with their consequences for both short- and long-term development
It is well established that maternal body status such as obesity and diabetes as well as dietary challenges such as over- or undernutrition and even maternal sickness can have profound effects on blastocyst morphogenesis across different species (Ashworth et al. 1999; Fleming et al. 2011; Williams et al. 2011; Fischer et al. 2012; Jungheim et al. 2012; Sinclair and Watkins 2013). For example, total cell number and the relative balance, allocation to and proliferation of ICM and TE are subject to plasticity in response to maternal dietary challenges (Ashworth et al. 1999; Kwong et al. 2000; Eckert et al. 2012). Equally, expression of genes involved in a large number of biological processes ranging from lineage differentiation to metabolic and epigenetic regulation can be influenced by maternal body status (Fleming et al. 2011). This is believed to be a compensatory response in an attempt to adapt and optimise development in prevailing conditions. But how does the early embryo sense such conditions and coordinate its responses to regulate growth? Mechanisms involved in transmitting external signals to the embryo thereby inducing appropriate responses are not fully understood but some key signalling networks are emerging and will be discussed below.
1.3.1 Insulin and Amino Acid Signalling—The mTORC Signalling Network
One example where adaptive responses of the developing embryo are starting to be better understood is the low protein diet (LPD) model in the mouse. In this model, offspring exposed to LPD exclusively during preimplantation development are more prone to hypertension, abnormal anxiety-related behaviour and obesity (Watkins et al. 2008). Reduced availability of insulin and amino acids (AAs) , specifically the branched-chain ones, is found around implantation following LPD (Eckert et al. 2012). Both insulin and AAs are well characterised to influence cell function and differentiation in early embryogenesis with long-lasting positive effects on fetal growth (Kaye and Harvey 1995; Heyner 1997; Lane and Gardner 1997; Martin and Sutherland 2001; Martin et al. 2003; Gonzalez et al. 2012). Insulin, for example, enhances mouse preimplantation embryo biosynthesis, proliferation and endocytosis (Dunglison et al. 1995; Kaye and Harvey 1995; Heyner 1997; Kaye and Gardner 1999). In conditions linked with insulin insufficiency such as maternal diabetes and hyperglycaemia early development is compromised (Pampfer 2000; Jungheim and Moley 2008). AAs are similarly beneficial for early embryo development with a role in biosynthesis and proliferation and as energy substrates as well as providing protective mechanisms against osmotic, metal ion and reactive oxygen stresses.
Both insulin and AAs instigate cellular responses using intracellular mTORC1 signalling (Proud 2007; Wang and Proud 2009, 2011; Dowling et al. 2010) to match nutrient availability. The mTORC1 serine-threonine kinase stimulates initiation of translation of cap-dependent and terminal oligopyrimidine (TOP)-dependent mRNAs through phosphorylation of two major downstream targets 4E-BP1 and S6 kinase (Proud 2007; Kim 2009; Wang and Proud 2009, 2011; Dowling et al. 2010). Whilst the extracellular or intracellular mechanisms for AA sensing and mTORC1 activation are not fully understood (Jewell et al. 2013), it is well-defined that insulin signals directly through the insulin receptor and IRS/PI3K/AKT/mTORC1 pathway (Cheng et al. 2010). Indeed, LPD can trigger a reduction in mTORC1 sensitivity and signaling through the S6 arm in blastocysts (Eckert et al. 2012). Therefore, it is plausible that the combined deficiencies in leucine, other branched chain AAs and insulin concentrations in the embryo environment following LPD function upstream as negative maternal factors in nutrient sensing by the embryo leading to developmental programming. However, overall protein biosynthesis is maintained in LPD blastocysts, possibly involving the 4E-BP arm of mTORC1 signalling which was not affected. Although mTORC1 signalling studies usually show both S6 and 4E-BP1 effectors to be responsive simultaneously, differential signalling disruption has been seen previously. More recent models of mTORC1 signalling suggest distinct biological roles and signalling pathways for the two main effectors S6 and 4E-BP1 (Duvel et al. 2010; Sengupta et al. 2010; Magnuson et al. 2012) as in the blastocysts exposed to maternal LPD. From a developmental point of view, such specific responses may be in place to protect developmental progress as the ultimate goal, by adapting embryonic metabolism and expenditure to prevailing conditions. In support of this view, implantation rates are not altered following maternal LPD, possibly due to a compensatory increase in TE proliferation and spreading ability around implantation (Watkins et al. 2008; Eckert et al. 2012). Experimental evidence suggests that signalling through the mTORC1 pathway plays an important role in trophoblast motility (Martin and Sutherland 2001; Martin et al. 2003; Gonzalez et al. 2012), proliferation (Kim et al. 2013) and implantation (Zeng et al. 2013) since embryos null for the mTOR gene arrest at E5.5 with implantation failure (Gangloff et al. 2004). In addition, the reduced availability of branched chain AAs following LPD acts to stimulate TE endocytosis through Rho A signalling, acting as a further mechanism of dietary compensation (Sun et al. 2014). Indeed, compensatory morphological organisation and transport activity in response to LPD in placentas in later gestation has been reported (Coan et al. 2011).
It is worth mentioning that despite the reduced maternal availability of branched chain AAs in uterine fluid following LPD and the accompanying reduction in blastocyst mTORC1 signalling, these AAs are present in blastocysts at near normal concentrations (Eckert et al. 2012). One could speculate that such distinction underpins the central role of these AAs in blastocyst metabolism and may be indicative of further compensatory alterations in uptake rate or transporter expression within blastocysts. For example, expression of the AA transporter SNAT-2 and other nutrient transporters in placenta are reduced in response to maternal LPD in later gestation (Jansson et al. 2006, 2012; Lager and Powell 2012) and mTORC1 signalling appears to be involved. It is suggested that nutrient sensing via mTORC1 may operate at cell surface or intracellular sites (Kim 2009; Taylor 2009; Jewell et al. 2013) thereby permitting concurrent mTORC1 signalling and compensatory activity. However, a more prominent role in such responses of mTORC2 which is deemed rapamycin-insensitive cannot be ruled out although mTORC2 components are normally decreased around blastocyst formation whilst it has recently been reported to be essential for early cleavage (Gonzalez et al. 2012; Zhang et al. 2012).
1.3.2 Energy Homeostasis—The AMPK Signalling Network
Models of insulin resistance and diabetes have similarly shown the impact of insulin signalling involving the mTORC pathway, yet in cross-talk with another key metabolic sensor, AMPK. AMPK is seen as the key responsible for maintaining cellular energy balance. When AMP levels rise, thus indicating ATP depletion, AMPK turns off ATP-consuming anabolic pathways including protein synthesis through mTORC1 inhibition and stimulates ATP-generating catabolic pathways such as glucose uptake and fatty acid oxidation via phosphorylation of downstream targets. None of the individual AMPK subunit deficiencies are lethal suggesting the ability to compensate for each other to a degree (Viollet et al. 2009). Evidence for a role of AMPK signalling in response to maternal status in early embryogenesis is indirect and stems from in vitro models (Eng et al. 2007; Louden et al. 2008) using embryo culture and trophoblast stem cells . When exposed to high insulin or IGF1 levels, blastocysts downregulate IGF1R thereby becoming insulin-resistant. Glucose uptake is reduced and apoptosis increased followed by early resorption and growth restriction. AMPK activators were able to reverse this effect by increasing the AMP/ATP ratio using trophoblast stem cells. Moreover, mTORC1 signaling through the S6K pathway was also increased, suggesting the cross-talk between these two energy sensing and expenditure pathways is operational in early development (Louden et al. 2008). Interestingly, downregulation of the IGF1R was not observed in in vitro derived bovine embryos exposed to high levels of IGF1 (Velazquez et al. 2011b). Nevertheless, downregulation of the IGF1R has been observed in blastocysts derived from obese cows (Velazquez et al. 2011a), a feature also reported in obese mice (Jungheim et al. 2010). It has been suggested that this downregulation of the IGF1R could be caused by the high levels of leptin usually present in obese individuals (Velazquez et al. 2011a).
Our knowledge about the role of AMPK signalling in early embryos is surprisingly scarce and partially conflicting (Bilodeau-Goeseels 2011). For example, whether AMPK activation inhibits or favours oocyte maturation remains somewhat controversial, may be species-dependent, and highly influenced by experimental conditions and the nature of AMPK activity modulators used (Bilodeau-Goeseels 2011). More recent data suggest a multifunctional role of AMPK which promotes meiotic maturation whilst inhibiting oocyte activation, at least in the mouse (Ya and Downs 2013). Possible explanations for our lack in understanding of its role in preimplantation development may be that the complex signalling network becomes fully functional only gradually, or the fact that AMPK signalling contributes to mediating stress-induced responses in early development (Xie et al. 2013). Most experimental data will involve varying levels of embryo stressors due to in vitro culture conditions. However, AMPK activation after osmotic stress in vitro can cause loss of potency factors such as Cdx2 and Id2 thereby influencing cellular differentiation . A similar mechanism may contribute to alterations in lineage allocations within the blastocyst seen as an early response to a variety of maternal dietary challenges (see above). Impairment of AMPK signalling certainly is a well-documented consequence observed in offspring in various organs including the heart and muscle (Gueant et al. 2013). To date, direct evidence for involvement of AMPK signalling triggering early embryonic responses to maternal diet is lacking. However, as discussed above, essential features of the maturing TE in blastocyst morphogenesis which are sensitive to environmental cues include tight junction formation and barrier function (Eckert and Fleming 2008) in concert with Na+/K+-ATPase activity (Armitage et al. 2008), collectively establishing cavitation as an interdependent process (Madan et al. 2007; Eckert and Fleming 2008; Giannatselis et al. 2011). Evidence from other systems suggests that AMPK signalling contributes to regulation of tight junction formation, barrier function and Na+/K+-ATPase activity directly or indirectly, possibly in response to short-chain fatty acids (Zhang et al. 2011; Benziane et al. 2012; Elamin et al. 2013) thus being a plausible target for transmitting responses to maternal diet in early embryos, too.
AMPK signalling is found upstream of some key cellular activities involving mitochondrial function and fatty acid metabolism, both of which are established targets of in utero exposure to environmental challenges. For example, mitochondrial dysfunction in mouse oocytes is seen as a consequence of maternal diet-induced obesity and insulin resistance or diets involving altered protein levels (Minge et al. 2008; Wakefield et al. 2008; Mitchell et al. 2009; Igosheva et al. 2010; Luzzo et al. 2012). In the mouse, studies using different maternal obesity models have shown altered mitochondrial structure and function, and increased potential, mitochondrial DNA content and biogenesis in oocytes and zygotes. This was accompanied by raised reactive oxygen species (ROS) and depleted glutathione resulting in a more oxidised redox state, suggestive of oxidative stress . Associated with these altered mitochondrial properties is the finding that a larger number of obese mothers fail to support blastocyst formation compared to lean dams (Igosheva et al. 2010) or growth retardation and abnormal brain development (Luzzo et al. 2012).
1.3.3 Lipid Metabolism and Fatty Acid Signalling
Another group of metabolic sensors linked to the AMPK signalling network are the transcription factors and members of the nuclear receptor superfamily, peroxisome proliferator-activated receptors (PPARs) (Feige et al. 2006). The three isoforms PPARα, PPARβ and PPARγ are encoded by separate genes and fulfil specific functions. They require activation by, for example, fatty acids and their derivatives, leukotrienes or prostaglandins and modulate expression of target genes in the cytoplasm or in the nucleus in response to ligand binding. Whilst it is well established that PPARs are key targets for adaptive processes even at the epigenetic level in offspring livers after exposure to maternal dietary challenge in utero in different species (Lillycrop et al. 2005; Altmann et al. 2013), their function in preimplantation embryo development is less well understood. Some evidence for a beneficial role in oocyte and embryo quality stems from an obesity mouse model. In an attempt to alleviate the accompanying insulin resistance using rosiglitazone, an activator of AMPK or PPARγ, blastocyst development was significantly improved and the increased blastomere allocation to the TE was normalised. This was not only coupled with maternal weight loss and improved glucose metabolism but with changes in ovarian mRNA expression of PPAR regulated genes, Cd36, Scarb1, and Fabp4 cholesterol transporters (Minge et al. 2008). Whilst it has long been established that PPARγ deficiency impairs final placental differentiation and causes lethality by day 10.5–11 (Barak et al. 1999; Kubota et al. 1999), it may also be a key target for metabolic regulation of ovarian function and oocyte and embryo quality early on. Mice deficient in either of the other two PPARs, α and δ, are viable and fertile showing relatively mild phenotypes (Lee et al. 1995; Peters et al. 2000) yet their role in blastocyst biogenesis has not been explored in detail. However, PPARδ, whilst apparently dispensable for blastocyst morphogenesis, can promote blastocyst hatching (Kang et al. 2011).
More recently, specific dietary treatments have been examined more closely with relation to oocyte quality and resulting embryo potential in farm animals. Maternal body composition, dietary carbohydrates and fatty acids, specifically polyunsaturated fatty acids, can impact on oocyte potential in cattle and sheep (Adamiak et al. 2006; Fouladi-Nashta et al. 2009; Wonnacott et al. 2010). Cattle commonly enter a state of negative energy balance after calving and the onset of lactation which can compromise immediate fertility. Elevated nonesterified fatty acid (NEFA) concentrations are commonly linked with this state as well as metabolic disorders such as obesity and type II diabetes. Dietary supplementation of rumen-protected conjugated linoleic acid (CLA) has been shown to help overcome fertility problems by increasing oocyte maturation and blastocyst development. This beneficial effect is linked to altered fatty acid composition and gene expression patterns of oocytes and embryos (Gonzalez-Serrano et al. 2013a). The development of techniques sufficiently sensitive to determine the fatty acid profile of individual oocytes and preimplantation embryos may help to elucidate signalling events involving fatty acids, their transporters and downstream metabolic pathways further after maternal dietary challenge (Gonzalez-Serrano et al. 2013b). In vitro exposure of oocytes to elevated NEFA levels during maturation impacts on gene expression and phenotype of the subsequent embryo including a disrupted oxidative metabolism. Expression of genes related to REDOX maintenance is modified and genes related to fatty acid synthesis are upregulated in NEFA-exposed oocytes, cumulus cells, and/or resultant blastocysts. In this model, inhibition of fatty acid β-oxidation in maturing oocytes exposed to elevated NEFA concentrations restored developmental competence although mitochondrial morphology and membrane potential remained unaltered unlike in rodent models exposed to maternal high fat diets (Igosheva et al. 2010; Luzzo et al. 2012) (discussed above). Collectively, these data suggest that mitochondrial function in fatty acid β-oxidation has a decisive impact on development, and that embryos adapt through altered metabolic strategies (Gonzalez-Serrano et al. 2013b). A growing body of evidence suggests that fatty acid signalling , specifically polyunsaturated fatty acids, can modify the epigenome. For example, expression of the fatty acid desaturase Fads2 in offspring liver is responsive to maternal intake of certain fatty acids during pregnancy in rats (reviewed in (Burdge and Lillycrop 2014; Gueant et al. 2014)). A variety of maternal diets (low protein, high fat, methyl donor deficient) and over-feeding leading to obesity appear to involve similar pathways to induce responses including the AMPK signalling network in its widest sense with some diets inducing very similar phenotypes in offspring (Gueant et al. 2014).
It is likely that the response of developmental and metabolic processes may be graded dependent upon the level of challenge by maternal body status. For example, a mouse model of dietary-induced obesity (Bermejo-Alvarez et al. 2012) confirms maternal obesity as a proxy for substantial endocrine and metabolic disruption linked to infertility at the ovarian level. However, a milder phenotype after the same dietary challenge was permissive for embryo survival to be near normal. Expression of key genes involved in metabolic signalling such as IGFR, adiponectin or leptin receptors remains unaffected in blastocysts whilst proteins involved in nutrient transport such as GLUT1 or LDLR, are downregulated. This may imply that metabolic plasticity of the preimplantation embryo initially aims at maintaining nutrient levels at manageable levels, at least in an overnutrition model (Bermejo-Alvarez et al. 2012). On the other hand, maternal hyperlipidaemia alone may be sufficient to induce impairment to fetal growth. Using a rabbit model (Picone et al. 2011; Cordier et al. 2013) a high fat diet did not induce weight gain due to appetite compensation but altered lipid metabolism, hormonal reproductive function and follicular growth in the mother and triggered abnormal placental vascularisation and IUGR in the fetus. Postnatally, offspring displayed increased adiposity, weight gain and hypertension similar to other rodent models mentioned above. Moreover, preimplantation embryos obtained from hyperlipidic dams may already adapt their lipid storage, overexpressing adipophilin or perilipin-2, a protein involved in lipid droplet formation and storage. The role of the perilipins in lipid handling is only beginning to emerge but may be important in determining the extent to which obesity may subsequently occur. Deficiency of perilipin-2 can protect against dietary-induced obesity and its consequences. Thus, its overexpression in early embryos as a consequence of a hyperlipidic environment may indicate a first sign of later weight gain and appetite dysregulation (Picone et al. 2011; Cordier et al. 2013; McManaman et al. 2013).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


