FIGURE 56-1 Implantable loop recorder in early exercise showing monomorphic premature ventricular contractions followed by salvos of polymorphic ventricular tachycardia.
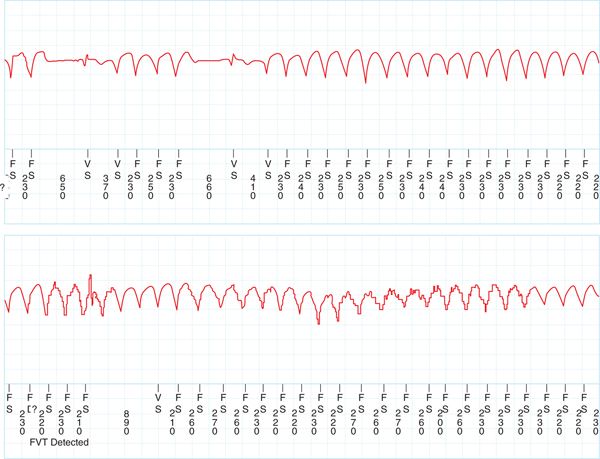
FIGURE 56-2 Implantable loop recorder showing sustained polymorphic ventricular tachycardia as exercise continues. This event was recorded as presyncope by the patient.
CASE EXPLANATION
Syncope during exercise is always concerning and requires a thorough evaluation. In patients with a structurally normal heart, the differential for a genetic electrical disorder includes the long QT syndromes, the short QT syndrome, Brugada syndrome, the Haissaguerre syndrome (idiopathic ventricular fibrillation associated with early repolarization), and CPVT. Of these syndromes, only CPVT has a normal baseline electrocardiogram with a normal QT interval. In addition to the baseline normal electrocardiogram, hallmarks of this syndrome include polymorphic or bidirectional VT that is reproducible with exercise and a family history of sudden death or syncope in an autosomal-dominant pattern. Based on recent investigations into the genetics of intracellular calcium regulation, the understanding of this disease has expanded, and this chapter will review the molecular genetics and discuss management of this newly described disease.
EPIDEMIOLOGY
• The first case report of CPVT was reported in 1975. A case series of four children with bidirectional ventricular tachycardia that was catecholamine-induced was reported in 1978.
• Multiple different cases series have now been reported, and currently it is estimated that the prevalence of the disease in 1:10,000 in Europe.1
• The syndrome is usually diagnosed in children and adolescents, although typically not before 2 years of age.
• The mortality of CPVT is 31% in untreated adults by the age of 30.1
PATHOPHYSIOLOGY
Calcium leakage from the sarcoplasmic reticulum (SR) is the mechanism of cellular cystolic calcium overload, resulting in CPVT. Normal calcium regulation during cardiac muscle contraction occurs in the SR and is controlled by the macromolecule called the calcium release complex (CRC)2 which is composed of the following:
The Cardiac Ryanodine Receptor (RyR2)3
• This is the main calcium release channel present in the SR.
• RyR2 is a large protein with a large cytoplasmic footprint that allows regulatory proteins to modulate its function.
• The channel works though calcium-induced calcium release.
 Calcium binds to RyR2 and triggers opening of L-type calcium channel, allowing rapid calcium to efflux from the SR.
Calcium binds to RyR2 and triggers opening of L-type calcium channel, allowing rapid calcium to efflux from the SR.
Calsequestrin (CASQ2)
• Calsequestrin is a large acidic protein that serves as a calcium buffer within the SR.
• Mechanistically, CASQ2 though interactions with triadin and junction confer RyR2 calcium luminal sensitivity such that CASQ2 inhibits RYR2 calcium release at low luminal calcium levels.4
Triadin and Junctin2
• Trisk 32 is an isoform of triadin found in cardiac muscle.
• Two isoforms of junctin are found in both skeletal muscle and cardiac muscle.
• Knockout mice of both Trisk 32 and junctin have resulted in fatal arrhythmias.
• These proteins anchor CSQ2 to RyR2 and work though a complex mechanism with CASQ2 to regulate SR calcium through the RYR2 membrane channel.
• Mutations in RyR2, CASQ2, and triadin lead to cystolic calcium overload, which generates delayed after depolarizations (DADs), triggered activity, and ventricular arrhythmias.
• Adrenergic stimulation results in an increased open probability of the RYR2 though phosphorylation of regulatory component of the RYR2, leading to higher levels of cytosolic calcium.
• In mouse models of CPVT, it appears that there is Purkinje origin of the ventricular premature beats resulting from calcium overload.5
ETIOLOGY
As our understanding of molecular genetics increases, mutations in the RyR2 membrane channel, calsequestrin, and tritan have been identified.
Mutations in RyR2
• RyR2 mutations are the most frequent mutations identified in CPVT.
• The typical pattern identified was found to be autosomal dominant and was found on chromosome 1q42-43.6
• RyR2 is a 4967-amino acid protein, one of the largest genes in the human genome, with 105 exons.
• At least 50 mutations have been identified in RYR2 that are likely associated with CPVT, although some mutations have been found in arrhythmogenic right ventricular cardiomyopathy.
• Most mutations occur in the N-terminal region, the central zone, or in the c-terminal region and are missense mutations.
• Despite having a disease-associated mutation, relatives carrying an RYR2 mutation have marked phenotypic variability, and no genetic genotype-phenotype correlations have been established.7
Mutations in CASQ2
• Mutations in CASQ2 are less common and have been described to map to chromosome 1p13-21.8
• CASQ2 is a 399 amino acid protein, and over 20 unique disease-causing mutations in this protein have been described.
• Approximately half the mutations are missense mutations, and the other half lead to premature stop codons, leading to a truncated protein.
• Phenotypically, patients with a single CASQ2 mutation have less severe symptoms than CPVT patients with RYR2 mutations, but patients with multiple mutations have been described and are susceptible to ventricular arrhythmias.
Mutations in Triadin2
• Three unique mutations of triadin have been identified in patients with clinical CPVT who do not have mutations in RYR2 or CASQ2. Two of these mutations resulted in a premature stop codon, and another led to a missense mutation.
• As some of these mutations affect all triadin isoforms, skeletal muscle could be affected, and muscle weakness has been observed in at least one patient with a triadin stop codon mutation.
Other Described Mutations
• A mutation on chromosome 7p14-p22 has also been described in an autosomal recessive Arab family with an early onset lethal form of CPVT, but no specific gene or protein in calcium regulation has been identified.9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


